
Identification of non-peptidic neuromedin U receptor modulators by a robust homogeneous screening assay1
Introduction
Neuromedin U (NMU) is a structurally, highly-conserved neuropeptide originally purified from the porcine spinal cord named for its uterine contractile property[1]. Two molecular forms with identical C-terminal octapeptide amides, NMU-8 and NMU-25, were characterized simultaneously and showed similar activities[2]. Several NMU analogs identified from different species display a high degree of conservation in amino acid sequences[3]. NMU has been implicated in a variety of physiological processes, including smooth muscle contraction[2], peripheral blood flow regulation and ion transport in the gut[4], food intake, energy homeostasis[5–7], stress responses[8], and pronociception[9]. Recently, its involvement in the pathogenesis of certain cancer and inflammation was demonstrated[10–13]. The action of NMU is mediated through 2 G-protein-coupled receptors (GPCR), NMU-R1 and NMU-R2. The former is expressed predominantly in peripheral tissues, and the latter is mainly located in the central nervous system[14–19]. Since specific roles of these 2 receptor subtypes are not fully understood, non-peptidic ligands could serve not only as potential drug leads, but also as powerful molecular probes to functionally characterize NMU receptors.
In the present study, we describe a robust scintillation proximity assay (SPA) to measure specific NMU-R1-binding properties of various ligands and its application in high-throughput screening (HTS) of 36 000 synthetic compounds or natural products. Three initial hits were discovered, and one of them (101586) was chosen as the scaffold for structural modifications and the subsequent structure–activity relationship (SAR) analyses. Of the 203 analogs synthesized, 16 retained variable human NMU receptor (hNMU-R) 1-binding abilities, but only 2 elicited calcium influx in both hNMU-R1 and hNMU-R2-expressing cells without observable antagonist activity.
Materials and methods
Reagents Sodium chloride, sodium phosphate dibasic anhydrous, potassium dihydrogen phosphate, potassium chloride, and EDTA were purchased from Shanghai Chemical Reagents (Shanghai, China). Bovine serum albumin (BSA) and fetal bovine serum (FBS) were procured from Sino-American Biotechnology (Shanghai, China) and Hyclone (Logan, UT, USA), respectively. Aprotinin, leupeptin, and G418 were bought from Merck KGaA (Darmstadt, Germany). The human NMU-25 peptide was obtained from Bachem AG (Bubendorf, Switzerland). [125I]hNMU-25 was made by ANAWA Trading SA (Wangen, Switzerland). FlashBlueTM GPCR beads and IsoplateTM mircotiter plates were the products of PerkinElmer (Boston, MA, USA). The FLIPR calcium 3 assay kit was supplied by Molecular Devices (Sunnyvale, CA, USA), probenecid by Sigma–Aldrich (St Louis, MO, USA), and cell culture medium F12 was from Invitrogen (Carlsbad, CA, USA).
Cell culture and membrane preparation Chinese hamster ovary cells (CHO-K1) stably expressing NMU-R1 or NMU-R2 were provided by Actelion Pharmaceuticals (Allschwil, Switzerland). They were maintained in F12 medium containing 10% FBS in the presence of G418 (400 mg/L) at 37 °C in a humidified atmosphere of 5% CO2. For the membrane preparation, the cells were treated with 0.25% trypsin (Sigma–Aldrich, USA) for 1 min and centrifuged at 1000×g for 10 min. The pellets were resuspended in the binding assay buffer (phosphate buffered saline with 1 mmol/L EDTA and 0.5% BSA, pH 7.4) and homogenized with a BioNeb® cell disruption system (Glas-Col, Terre Haute, IN, USA) followed by centrifugation at 1200×g at 4 °C for 20 min to precipitate debris. The supernatant was centrifuged at 17 000×g for 30 min to pellet the membrane that was resuspended in the binding assay buffer thereafter. The protein content was determined using a spectrophotometer (Thermo Electron, Waltham, MA, USA).
SPA-binding assay Various amounts of the above membrane receptor preparations, 0.04 nmol/L [125I]hNMU-25, FlashBlueTM GPCR beads (100 µg/well), different concentrations of non-labeled hNMU-25, aprotinin (5 µg/mL), and leupeptin (5 µg/mL) were added to the binding assay buffer to give a final volume of 0.1 mL. The plates were incubated at room temperature for 3–4 h before counting on the Microbeta scintillation counter (PerkinElmer, USA).
Calcium mobilization assay The above cells were detached and plated onto 96-well clear culture plates (Corning, Acton, MA, USA) at a density of 30 000–40 000 cells (100 µL/well) and incubated overnight. The cells were loaded with 100 µL of the calcium 3 assay dye supplemented with 2.5 mmol/L probenecid, and incubated at 37°C for 60 min. A baseline fluorescence signal was measured for the first 17 s, after which the test compounds prepared as stock with different concentrations (1 mmol/L–3 µmol/L) in Hanks’ balanced salt solution buffer (supplied with the assay kit) were added to the plate through an automated pipetter (20 µL/well) equipped within FlexStation II384 (Molecular Devices, USA). The intracellular calcium influx was analyzed by the same instrument with an excitation wavelength of 485 nm and emission wavelength of 525 nm; the relative fluorescence signal was measured at 1.6 s intervals for 150–300 s. hNMU-25 was used as a positive control in this homogeneous fluorescence emission assay.
HTS campaign The compound library used for the screening consisted of 20 000 pure synthetic compounds and 16 000 natural products. A 10-compound pool per well mix was applied to the primary screening, with an average concentration of 7 µmol/L for each compound dissolved in 100% DMSO solution. This matrix system maximizes the advantage of HTS and allows duplicate screening of each compound[20]. In each 96-well IsoplateTM, 14 wells were used as positive controls (hNMU-25) and 2 wells as negative controls (2.5% DMSO alone). Samples showing greater than 30% inhibition were considered “hits”.
Chemistry Reagents were obtained from Lancaster (Morecambe, England), Aldrich (St Louis, MO, USA), Acros (Geel, Belgium), and Shanghai Chemical Reagents (China). All of the reagents were of analytical pure grade or above and were used without further purification. The analytical thin-layer chromatography was performed on HSGF 254 silica gel (150–200 µm thickness; Yantai Huiyou, Yantai, Shandong, China). Column chromatography was performed using 200–300 mesh silica gels (Qingdao Haiyang Chemical, Qingdao, Shandong, China). Yields were not optimized. Melting points were recorded on a capillary tube on a SGW X-4 melting point apparatus without correction (Shanghai Precision and Scientific Instrument, Shanghai, China). 1HNMR was recorded in d-chloroform on a Varian Mercury 300 or 400 (300 or 400 MHz) NMR spectrometer (Varian, Fort Collins, CO, USA). Chemical shifts were reported in parts per million (ppm, δ). Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low-resolution mass spectra (LRMS) were measured by the electric ionization (EI) method with a Finnigan MAT-95 (Finnigan, Santa Clara, CA, USA). The Liquid chromatography-electrospray ionization mass (ESI) was carried out on a Thermo Finnigan LCQDECAXP (Thermo Finnigan, San Jose, CA, USA).
The general preparation procedure for compounds 1a–1d is illustrated in Figure 1. 1,8-Diazabicyclo(5.4.0)undec-7-ene (2.8 g, 18.2 mmol) and tert-butyldiphenylchlorosilane (2.2 g, 36.3 mmol) were added to 2-aminoethanol (5.0 g, 18.2 mmol) in 50 mL of acetonitrile, and reacted overnight. The solvent was evaporated in vacuo, and the residues were extracted with ethyl acetate, washed with water/brine, and dried over anhydrous Na2SO4. Evaporation of the solvent gave 5.2 g of crude product 6a (yield: 95.5%). It was subsequently dissolved (100 mg, 0.33 mmol) in 2 mL acetonitrile followed by the addition of 2-(methylthio)-4,5-dihydro-1H-imidazole hydroiodide (81.5 mg, 0.33 mmol). The resulting mixture was heated to reflux for 8 h, and evaporation of the solvent in vacuo delivered a crude product that was purified by silica gel chromatography to afford 78 mg of compound 1a (yield: 64%) as a white amorphous solid. The structure was determined with 1HNMR (400 MHz, CDCl3) δ 8.43 (br, 1H), 7.96 (br, 1H), 7.40–7.64 (m, 10H), 6.7 (br, 1H), 3.88 (t, J=4.5 Hz, 2H), 3.74 (m, 2H), 3.44 (m, 4H), 1.09 (s, 9H); and with LRMS (EI, 70 eV): m/z 367 [M+], 310 (100%). Compounds 1b–1d were prepared accordingly. The structure of compound 1b (88.2 mg; yield: 69.2%) was determined with 1HNMR (400 MHz, CDCl3) δ 7.99 (br, 1H), 7.41–7.51 (m, 10H), 4.12 (t, J=4.1 Hz, 2H), 3.36 (m, 2H), 3.23 (m, 4H), 1.88 (m, 2H), 1.09 (s, 9H); and with LRMS (EI, 70 eV): m/z 381 [M+], 310 (100%). The structure of compound 1c (79 mg; yield: 65%) was determined with 1HNMR (400 MHz, CDCl3) δ 7.92 (br, 1H), 7.58–7.62 (m, 10H), 7.0 (br, 1H), 3.81 (t, J=5.5 Hz, 2H), 3.73 (t, J=8.3 Hz, 2H), 3.46 (m, 4H), 1.74 (m, 2H), 1.09 (s, 9H); and with LRMS (EI, 70 eV): m/z 381 [M+], 310 (100%). The structure of compound 1d (83.2 mg; yield: 65.9%) was determined with 1HNMR (400 MHz, CDCl3) δ 7.78 (br, 1H), 7.42–7.60 (m, 10H), 3.84 (t, J=5.4 Hz, 2H), 3.39 (m, 2H), 3.19 (m, 4H), 1.90 (m, 2H), 1.1 (s, 9H); and with LRMS (EI, 70 eV): m/z 395 [M+], 310 (100%).
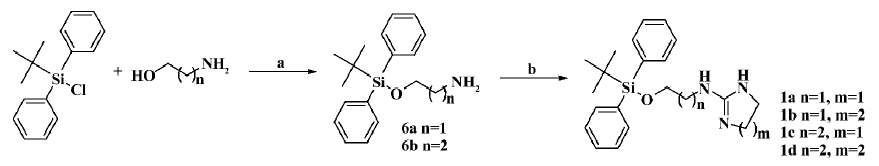
The general preparation procedure for compounds 2a–2d is illustrated in Figure 2. TsOH (1.0 g, 5.43 mmol) was added to diphenylmethanol (10.0 g, 54.3 mmol) and 2-bromoethanol (13.6 g, 108.6 mmol) in 50 mL anhydrous toluene solution, and the mixture heated to 60oC for 8 h. Following the addition of 10 mL saturated NaHCO3 aqueous solution, the toluene layer was separated and washed with water and saturated NaCl aqueous solution. It was then dried over Na2SO4 and evaporation of the solvent in vacuo gave 14.9 g of compound 7a as a colorless oil (yield: 94.3%). Compound 7a (10.0 g, 34.3 mmol) was dissolved in 40 mL N,N-dimethylformamide with the addition of 17.9 g (0.27 mol) NaN3 in an ambient temperature for reaction at 70oC for 3 h. The mixture was then quenched into 100 mL water and the aqueous phase was extracted with 50 mL ether 3 times. Combining the organic layer, washing with water and saturated NaCl aqueous solution, drying over Na2SO4, and removing the solvent in vacuo afforded compound 9a as pale yellow oil. It was subsequently dissolved (100 mg, 0.44 mmol) in 2 mL acetonitrile and mixed with 2-(methylthio)-4,5-dihydro-1H-imidazole hydroiodide (107 mg, 0.44 mmol). Heating to reflux for 8 h and evaporation of the solvent in vacuo delivered a crude product, which was purified by silica gel chromatography to give compound 2a (93 mg; yield: 72%) as a white amorphous solid. The structure was determined with 1HNMR (300 MHz, CDCl3) δ 8.36 (br, 1H), 7.79 (br, 1H), 7.27–7.38 (m, 10H), 6.81 (br, 1H), 5.44 (s, 1H), 3.69 (m, 2H), 3.65 (br, 2H), 3.49 (m, 2H), 3.26 (br, 2H); and with ESI: m/z (relative intensity) 296 (M+1, 100%), 167. Compounds 2b–2d were made the same. The structure of compound 2b (92 mg; yield: 67%) was determined with 1HNMR (300 MHz, CDCl3) δ 7.82 (t, J=6.0 Hz, 1H), 7.63 (br, 1H), 7.27–7.38 (m, 10H), 5.44 (s, 1H), 3.64 (m, 2H), 3.41 (m, 2H), 3.08 (br, 4H), 1.77 (m, 2H); and with ESI: m/z (relative intensity) 310 (M+1, 100%), 167. The structure of compound 2c (82 mg; yield: 64%) was determined with 1HNMR (300 MHz, CDCl3) δ 7.27–7.35 (m, 10H), 5.40 (s, 1H), 3.60 (m, 2H), 3.56 (br, 2H), 3.42 (m, 2H), 3.20 (br, 2H), 1.92 (m, 2H); and with ESI: m/z (relative intensity) 310 (M+1, 100%), 167. The structure of compound 2d (102 mg; yield: 75%) was determined with 1HNMR (300 MHz, CDCl3) δ 7.59 (br, 1H), 7.28–7.36 (m, 10H), 5.38 (s, 1H), 3.60 (t, J=5.6 Hz, 2H), 3.36 (dd, J=6.7, 12.6 Hz, 2H), 2.95 (br, 4H), 1.38 (m, 2H), 1.75 (m, 2H); and with ESI: m/z (relative intensity) 324 (M+1, 100%), 167.
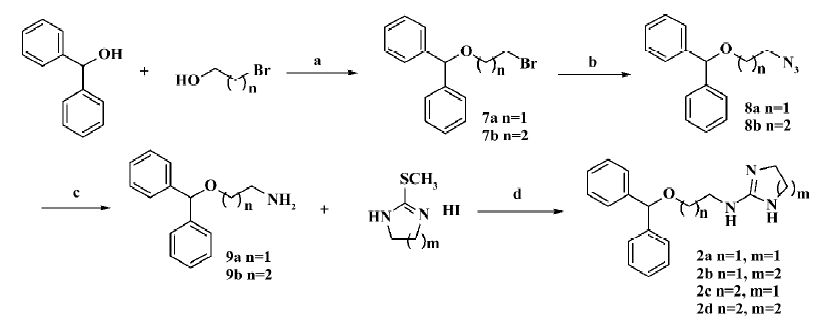
The general preparation procedure for compounds 3a–3d is illustrated in Figure 3. Tert-butyl 2-aminoethylcarbamate (1.4 g, 8.63 mmol in 10 mL dichloromethane) was added to 2.0 g (8.63 mmol) diphenylcarbamic chloride in 10 mL CH2Cl2 (cooled to 0oC) in a drop-wise manner. N,N’-diisopropylethylamine (1.1 g, 8.63 mmol) and 4-dimethylaminopyridine (105 mg, 0.86 mmol) were introduced thereafter. The stirred reaction was carried out at room temperature overnight followed by dilution with an extra 30 mL dichloromethane. The solution was then sequentially washed with water, 1 mol/L HCl aqueous solution and saturated NaCl aqueous solution, dried over Na2SO4, and the solvent removed in vacuo. The resultant product 10a was recrystallized from petroleum ether/EtOAc as a white solid (2.6 g; yield: 85%). Compound 10a was dissolved in a mixture of 10 mL trifluoroacetic acid and 10 mL dichloromethane and stirred at room temperature for 0.5 h. It was poured into a 50 mL saturated Na2CO3 aqueous solution and extracted with 100 mL dichloromethane. The organic layer was washed with water and the saturated NaCl aqueous solution, dried over Na2SO4, and the solvent evaporated in vacuo to give crude product 11a for use without further purification. Compound 3a (a white amorphous solid) was similarly prepared as compound 1a (79 mg; yield: 62%). The structure was determined with 1HNMR (400 MHz, CDCl3) δ 8.38 (br, 1H), 8.28 (t, J=6.3Hz, 1H), 7.26–7.45 (m, 10H), 3.69 (s, 4H), 3.42 (m, 2H), 3.31 (m, 2H); and with ESI: m/z (relative intensity) 324.2 (M+1, 100%), 239, 155. Compounds 3b–3d were synthesized accordingly. The structure of compound 3b (a white solid, 88 mg; yield: 67%; melting point: 120–124oC) was determined with 1HNMR (300 MHz, CDCl3) δ 7.66 (br, 1H), 7.59 (m, 1H), 7.23–7.42 (m, 10H), 3.56 (m, 2H), 3.30 (m, 2H), 3.21 (m, 4H), 1.84 (m, 2H); and with ESI: m/z (relative intensity) 338 (M+1, 100%), 239, 169. The structure of compound 3c (a white amorphous solid, 72 mg; yield: 57%) was determined with 1HNMR (300 MHz, CDCl3) δ 8.23 (br, 1H), 7.78 (br, 1H), 7.20–7.40 (m, 10H), 6.99 (m, 1H), 3.46 (m, 4H), 3.37 (m, 2H), 3.27 (m, 2H), 1.78 (m, 2H); and with ESI: m/z (relative intensity) 338 (M+1, 100%), 253, 169. The structure of compound 3d (a white solid, 76 mg; yield: 60%; melting point: 195–198oC) was determined wuth 1HNMR (300 MHz, CDCl3) δ 8.08 (t, J=6.0 Hz, 1H), 7.56 (dd, J=1.2, 7.8 Hz, 2H), 7.43 (dd, J=1.4, 4.3 Hz, 2H), 7.39 (td, J=7.7 Hz, 1.4 Hz, 2H), 7.26 (td, J=7.6, 1.3 Hz, 2H), 7.16 (br, 1H), 5.85 (t, J=5.5 Hz, 1H), 3.64 (s, 4H), 3.41–3.47 (m, 2H), 3.31–3.37 (m, 2H); and with ESI: m/z (relative intensity) 354.1 (M+1, 100%).
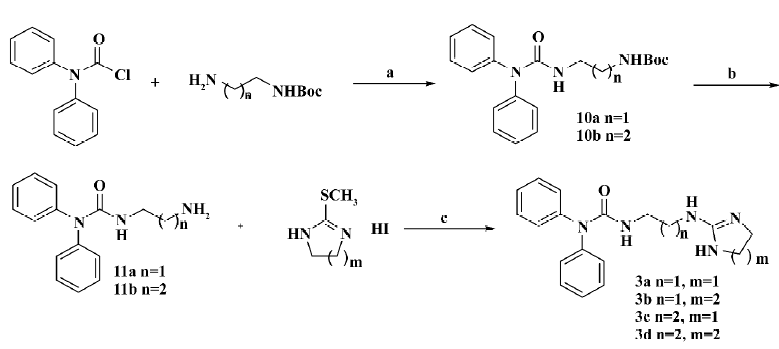
The general preparation procedure for compounds 4a–4c and 5a–5b is illustrated in Figure 4. Trityl chloride (3.6 g, 0.015 mol) was added to a solution of N-hydroxylphthalimide in 40 mL dry CH2Cl2 and N,N’-diisopropylethylamine (2.8 mL, 0.015 mol). The completion of the reaction was confirmed by thin layer chromatography (TLC) after 20 h. Hydrazine hydrate (85%, 16 mL) was then introduced to the mixture, followed by the addition of methanol until it became a homogenous solution. Upon confirmation by TLC after 1 h, the completed reaction mixture was diluted with CH2Cl2, washed with NaHCO3 aqueous solution, and dried over Na2SO4. The residue was applied to a silica gel column, eluted with CH2Cl2:MeOH=50:1, and resulted in 2.49 g of compound 12a. It was dissolved (100 mg, 0.33 mmol) in 2 mL acetonitrile before adding 2-(methylthio)-4,5-dihydro-1H-imidazole hydroiodide (85 mg, 0.33 mmol). The mixture was then heated to reflux for 8 h and the solvent evaporated in vacuo to deliver a crude product; the subsequent purification by silica gel chromatography afforded 87.3 mg of compound 4a (yield: 71%) as a pale yellow solid, melting point: 108~110oC. Its structure was confirmed with 1HNMR (300 MHz, CDCl3) δ 8.33 (br, 1H), 7.93 (br, 1H), 7.24–7.38 (m, 15H), 6.97 (br, 1H), 3.70 (m, 2H), 3.45 (m, 4H), 3.37 (m, 2H); and with ESI: m/z (relative intensity) 372.2 (M+1, 100%), 243.1. Compounds 4b–4c were prepared accordingly. The structure of compound 4b (a pale yellow solid, 93 mg; yield: 76%; melting point: 160–163oC); was determined with 1HNMR (300 MHz, CDCl3) δ 8.08 (br, 1H), 7.61 (br, 1H), 7.23–7.37 (m, 15H), 6.59 (br, 1H), 3.56 (m, 2H), 3.45 (m, 2H), 3.25 (m, 4H), 1.86 (m, 2H); and with ESI: m/z (relative intensity) 386.1 (M+1, 100%), 243.1. The structure of compound 4c (amorphous yellow solid, 92 mg, 76%) was determined with 1HNMR (300 MHz, CDCl3) δ 7.25–7.39 (m, 15H), 3.58 (m, 4H), 3.09 (m, 2H), 2.98 (m, 2H), 1.56 (m, 4H); and with ESI: m/z (relative intensity) 400.2 (M+1, 100%), 243.1. Compound 5a was made from 12a (200 mg, 0.66 mmol), which was dissolved in 1 mL N,N-dimethylformamide prior to the addition of 1H-pyrazole-1-carboxamidine hydrochloride (0.11 g, 0.73 mmol) and N,N’-diisopropylethylamine (0.12 mL, 0.73 mmol). Ether (5 mL) was added to the reaction while stirring at room temperature, and the crude product was precipitated as the oil. The ether layer was then poured out, and the residue purified by silica gel chromatography afforded 136 mg of compound 5a (yield: 60%) as a white solid, melting point: 189–193oC. Its structure was determined with 1HNMR (300 MHz, CDCl3) δ 7.23–7.41 (m, 15H), 3.32 (m, 2H), 2.98 (m, 2H); and with ESI: m/z (relative intensity) 346.1(M+1, 100%), 243.1. Compound 5b was prepared similarly as a white solid (156 mg; yield: 69%), melting point: 148–151oC. The structure was determined with 1HNMR (300 MHz, CDCl3) δ 7.19–7.39 (m, 15H), 3.11 (m, 2H), 2.99 (m, 2H), 1.77 (m, 2H); and with ESI: m/z (relative intensity) 360.2 (M+1, 100%), 243.1.

Data analysis Data were analyzed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Non-linear regression analyses were performed to generate dose-response curves to calculate IC50 (the molar concentration of an antagonist, which produces 50% of maximum possible inhibition of radiolabeled ligand binding for that antagonist in the receptor-binding assay) or EC50 (the molar concentration of an agonist, which produces 50% of the maximum possible response for that agonist) values. Ki values were calculated from IC50 using the equation of Cheng and Prusoff[21] (see below). The time-course study results from the calcium mobilization assay were expressed as the relative fluorescence unit (RFU), and all other calcium influx measurements were represented as the peak RFU per well. Values presented are mean±SEM of at least 3 independent experiments. Z’ factors were calculated as described by Zhang et al[22].
Results
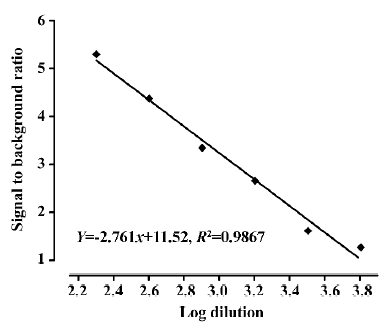
Assay validation We first used various concentrations of the hNMU-R1 membrane extract to assess the optimal protein concentration for the SPA assay. Both maximum binding (MB; using DMSO) and non-specific binding (NSB; using 1 µmol/L hNMU-25) were investigated, and the optimal hNMU-R1 concentration was determined to be 2 µg/well (1:230 of the stock solution) that resulted in a signal to background (S/B) ratio of 5 (Figure 5). To confer HTS requirements, other assay parameters, such as the quantity of [125I]hNMU-25, amount of beads, and reaction volume were also optimized (Figure 6).
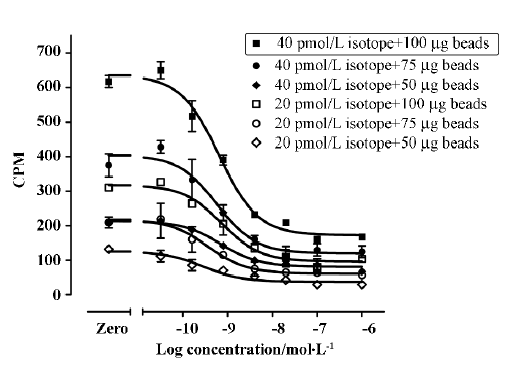
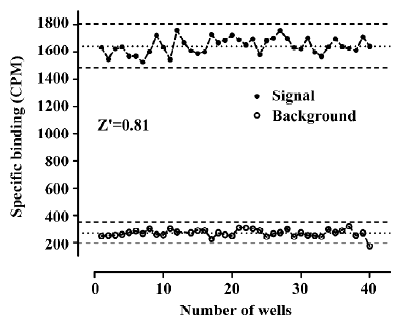
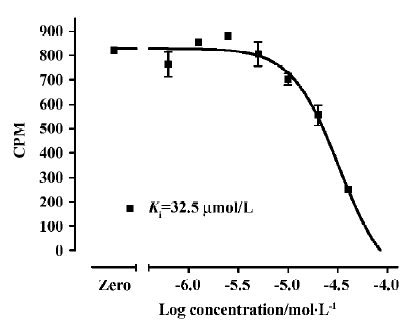
Assay performance As shown in Figure 7, the average Z' value for the SPA assay was 0.81 with a S/B ratio of 6, suggesting that the system was adequately optimized for HTS. Assay stability was evaluated by incubating the plates overnight at room temperature and both the Z' factor and S/B ratio remained unchanged (data not shown). The coefficient of variation (CV) values were 3.6% for MB and 10.1% for NSB, respectively. The known hNMU-R1 ligand hNMU-25 was used to verify the methodology, and the binding affinity (Ki) observed was consistent with that reported in the literature (Figure 8)[5].

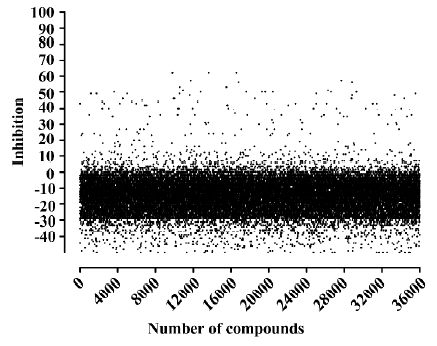
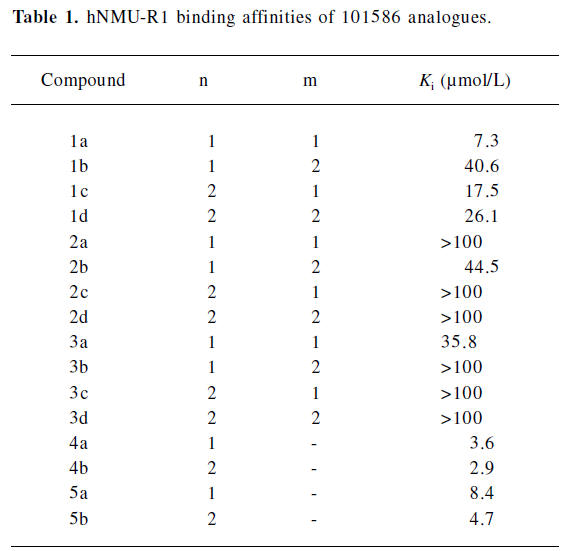
HTS campaign Of the 36 000 samples screened, 100 hits (0.28%) showing greater than 30% competitive inhibition on [125I]hNMU-25 binding to hNMU-R1 were discovered (Figure 9). Secondary (single compound per well) screening confirmed that 3 of the above hits displayed consistent inhibitory effects with Ki values around 30 µmol/L. All were synthetic chemicals with similar structures and newly identified ligands for hNMU-R1. Compound 101586 was chosen as the scaffold for the structural modifications (Figures 10, 11).
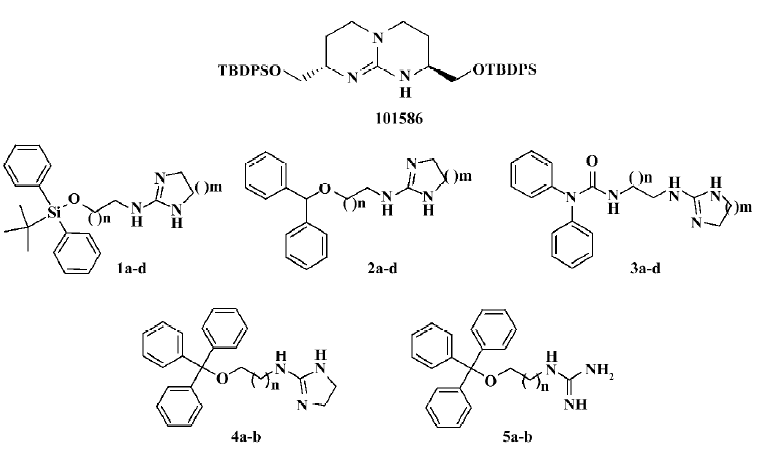
Structural modifications Five groups of analogs to 101586 were designed and synthesized. Compounds 1a–d were designed based on the SAR information relative to porcine NMU-8[23–29]. The diphenyl group was introduced to resemble the hydrophobic residues in this peptide, as we believe this group may bind to a hydrophobic site on the hNMU-R, and that an interaction with an anionic site of the receptor might be realized by linking to a cyclized guanidine group with changes in the carbon chain length and ring size. Such an attempt (substitution with a diphenyl group) yielded compounds 1a–d and one of them (1a) exhibited an improved binding affinity to hNMU-R1 (Ki=7.30 µmol/L; Table 1, Figure 11). When the protective group tert-butyldiphenylsilyl was substituted by diphenylmethyloxy (2a–d) or diphenylurea group (3a–d) to remove potential toxicity, only 2b and 3a showed weak hNMU-R1-binding ability, suggesting that the extra tributyl in the tert-butyldiphenylsilyl group is required for receptor binding. Thus, compounds 4a–b and 5a–b were prepared by adding a phenyl group onto the diphenylmethyl site in order to enhance their binding capability. As a result, these 4 analogs displayed significant improvements in hNMU-R1-binding affinities, with Ki values ranging between 2.9 and 8.4 µmol/L (Table 1; Figure 11). We also replaced the guanidine group with 2-(thiophen-2-yl)ethanyl, 2-(pyridin-4-yl)ethanyl, 3-(1H-imidazol-1-yl)propan-1-yl, 2-(2-chlorophenyl)ethanyl, 4-phenylbutan-2-yl, 2-phenylethanyl, or octan-1-yl, 2-cyclohexenylethanyl groups, but none of the substituted compounds showed observable bioactivities (data not shown). The above limited SAR study led us to postulate that the oxygen atom at the triphenylmethoxyl group seems important to NMU-R1 binding, while the tritylthio or tritylamino groups do not have this property.
Full table
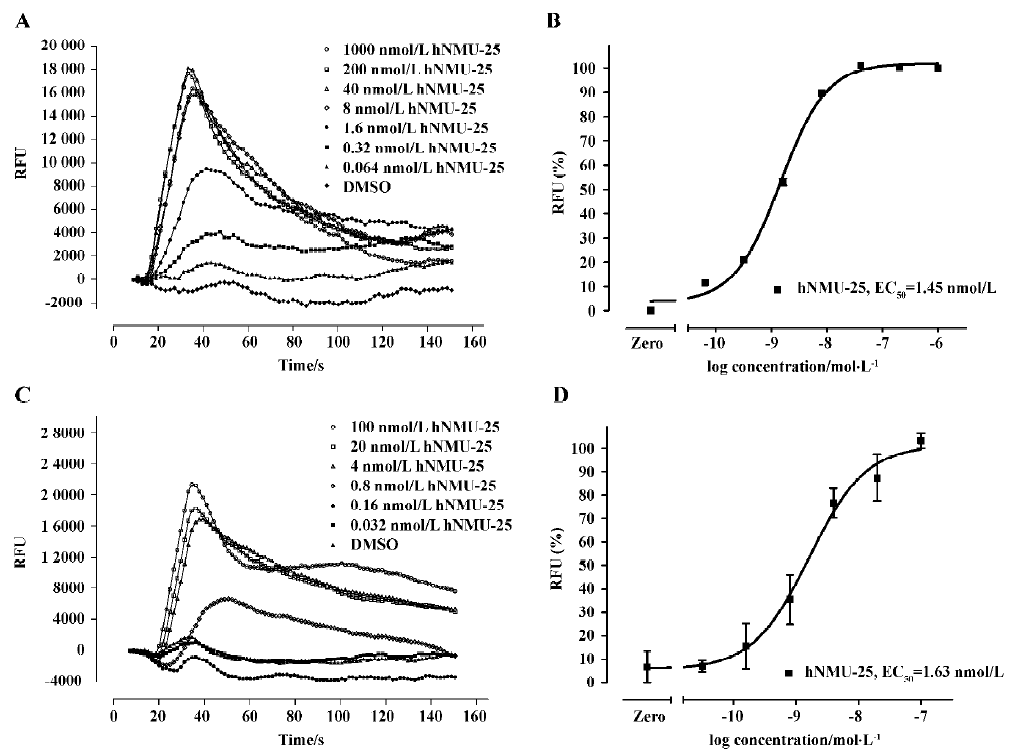
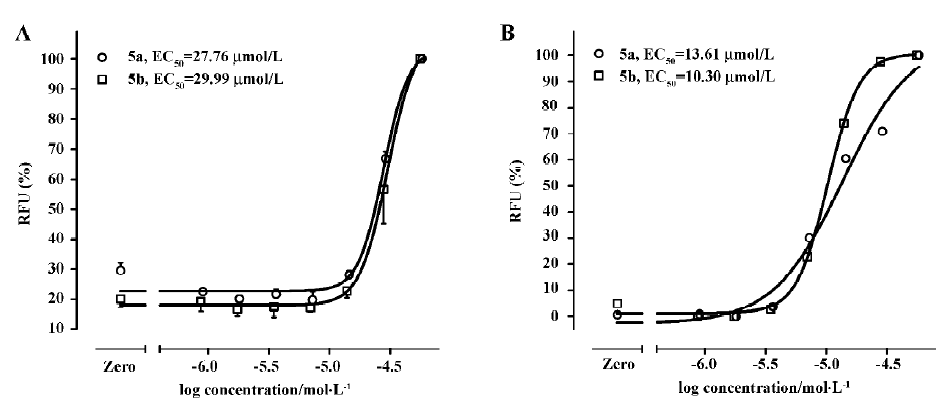
Functional analysis The pharmacological property (agonist or antagonist) was evaluated with a calcium mobilization assay using hNMU-R1 and hNMU-R2-expressing cells, respectively. Figure 12 depicts the kinetics and concentration-dependent agonist effects of hNMU-25 on calcium influx in these 2 types of engineered cells. The EC50 values estimated from the regression curves were 1.45 nmol/L (hNMU-R1 cells) and 1.63 nmol/L (hNMU-R2 cells), respectively, consistent with those reported in the literature[5]. Of the 16 analogs to 101586, 10 possessing Ki values below 50 µmol/L were functionally tested. Among them, only 5a and 5b demonstrated moderate and non-selective agonist activities in both hNMU-R1 (Figure 13A) and hNMU-R2 cells (Figure 13B). None of the analogs showed any antagonistic effects (data not shown).
Discussion
Due to their large variety of biological activities, studies on NMU and its cognate receptors have become an emerging area that attracts scientists from different disciplines. Apart from the potential value of using NMU-R1 and/or NMU-R2 as drug targets, exploration of small molecule ligands for these receptors could provide powerful tools in further understanding the physiological roles of this peptide family. Here we report a rational approach for discovering novel and non-peptidic modulators for NMU receptors, combining both molecular and cell-based functional assays.
Taking advantage of our prior experience in the development of SPA-based, receptor-binding HTS methods[30,31], efforts were made to expand the knowledge to NMU-R. A number of assay parameters were sequentially evaluated and optimized to enhance the S/B ratio. It is well recognized from other membrane receptor screening settings that assay sensitivity can be improved by choosing a radiolabeled ligand concentration at or below its Kd to permit effective competition by an unlabeled ligand[32]. In the present study, therefore, we selected 0.04 nmol/L as the concentration for [125I] hNMU-25, which is well below the Kd value (0.3 nmol/L) of unlabeled ligands[5]. A stable signal is highly dependent on protein stability and its coating ability on the beads. In our assay system, no signal shift was observed for more than 48 h (data not shown), implying a very stable interaction among various reagents.
The Z' factor is a useful indicator for assessing the quality of HTS assays[22]. In general, a Z' value above 0.5 suggests that an assay is robust enough for use in HTS settings. The SPA system described in this paper consistently displayed a Z' value of 0.81. This, and in conjunction with other parameters, such as the S/B ratio and CV values, suggests that the assay is of high-quality nature. When reduced to practice, it led to the discovery of 3 confirmed hits that bind to NMU-R1 in a high micromolar range (~30 µmol/L).
According to the common molecular skeleton represented by compound 101586 (as a molecular recognition chiral building block[33]), 203 analogs were synthesized and tested. Among the 16 analogs that retained variable hNMU-R1-binding abilities, 2 (5a and 5b) elicited calcium influx in both hNMU-R1 and hNMU-R2-expressing cells but none displayed antagonist activity. Our structural modifications were carried out based on the SAR analysis of peptidic NMU analogs, especially for NMU-8. It is known that these structurally highly conserved neuropeptides share a motif of FLFRPRX-amide, suggesting the importance of this sequence in the interaction with its cognate receptors. Specifically, NMU-8 peptides precede with a large flexibility of the 4 amino acids sequence rich in hydrophobic residues, namely Tyr1(or Phe1)–Phe2–Leu3(or Val3)–Phe4. This sequence is succeeded by a hydrophilic and base C-terminal Arg5–Pro6–Arg7–Asn8-NH2 in all species, implying that an amphiphilic structure could be designed[23–29]. Diphenyl groups are widely present in many drugs on the market targeting GPCR, including adrenergic receptor antagonists, muscarinic receptor antagonists, analgesic agents (opioid receptor agonists), anti-allergic medications (histamine receptor-1 antagonists), and serotonin receptor modulators[34]. The scaffold of these GPCR modulators can be summarized as the template in Figure 14.
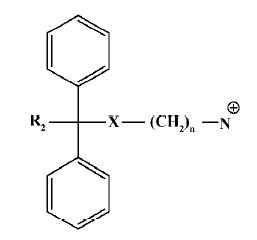
Indeed, when the diphenyl group was introduced to resemble the hydrophobic residues in NMU-8, compound 1a showed an improved binding affinity to hNMU-R1; the addition of a phenyl group onto the TBDPS' diphenylmethyl site also increased the receptor-binding capability (compounds 4a–b and 5a–b); the substitution of TBDPS with diphenylmethyloxy (2a–d) or the diphenylurea group (3a–d), and replacement of the guanidine group with other functional groups did not yield satisfactory results. It is thus clear that the oxygen atom at the triphenylmethoxyl group is required for NMU-R1 binding that does not involve the tritylthio or tritylamino groups. This preliminary SAR investigation further revealed some common structural features of this class of 101586 analogs necessary for the observed in vitro activities: the distance between the hydrophobic part and cyclized guanidine should not exceed 3 carbons in length, a 5-membered ring is better than a 6-membered ring in the cyclic guanidines, and the isolated guanidine was indispensable for hNMU-R activation.
In summary, a simple and homogeneous SPA-based, HTS-binding assay was developed and validated for the identification of novel non-peptidic ligands with specificity and functionality for NMU receptors. The newly-discovered NMU-R modulators have distinct structural features compared to that reported elsewhere[35]. Knowledge obtained from the present study will certainly facilitate the pursuit of developing small molecule agonists and/or antagonists directed at hNMU-R with better potency and efficacy.
Acknowledgements
We are indebted to Mr Jie GAO and Ms Xi-yuan CHENG for their technical assistance, and to Dr Dale MAIS for critical review of this manuscript.
References
- Minamino N, Sudoh T, Kangawa K, Matsuo H. Neuromedins: novel smooth-muscle stimulating peptides identified in porcine spinal cord. Peptides 1985;6 Suppl 3:245-8.
- Minamino N, Kangawa K, Matsuo H. Neuromedin U-8 and neuromedin U-25: novel uterus stimulating and hypertensive peptides identified in porcine spinal-cord. Biochem Biophys Res Commun 1985;130:1078-85.
- Brighton PJ, Szekeres PG, Willars GB. Neuromedin U and its receptors: structure, function and physiological roles. Pharmacol Rev 2004;56:231-48.
- Sumi S, Inoue K, Kogire M, Doi R, Takaori K, Tobe T, et al. Effect of synthetic neuromedin U-8 and U-25, novel peptides identified in porcine spinal-cord, on splanchnic circulation in dogs. Life Sci 1987;41:1585-90.
- Howard AD, Wang R, Pong SS, Mellin TN, Strack A, Liu Q, et al. Identification of receptors for neuromedin U and its role in feeding. Nature 2000;406:70-4.
- Nakazato M, Hanada R, Murakami N, Date Y, Mondal MS, Matsukura S, et al. Central effects of neuromedin U in the regulation of energy homeostasis. Biochem Biophys Res Commun 2000;277:191-4.
- Ivanov TR, Lawrence CB, Stanley PJ, Luckman SM. Evaluation of neuromedin U actions in energy homeostasis and pituitary function. Endocrinology 2002;143:3813-21.
- Hanada R, Nakazato M, Murakami N, Sakihara S, Yoshimatsu H, Sakata T, et al. A role for neuromedin U in stress response. Biochem Biophys Res Commun 2001;289:225-8.
- Yu XH, Cao CQ, Mennicken F, Puma C, Dray A, Perkins M, et al. Pro-nociceptive effects of neuromedin U in rat. Neuroscience 2003;120:467-74.
- Alevizos I, Mahadevappa M, Zhang X, Ohyama H, Kohno Y, Wong DT, et al. Oral cancer in vivo gene expression profiling assisted by laser capture microdissection and microarray analysis. Oncogene 2001;20:6196-204.
- Shetzline SE, Rallapalli R, Dowd KJ, Zou SM, Nakata Y, Gewirtz AM, et al. Neuromedin U: a Myb-regulated autocrine growth factor for human myeloid leukemias. Blood 2004;104:1833-40.
- Wu Y, McRoberts K, Berr SS, Frierson HF Jr, Conaway M, Theodorescu D. Neuromedin U is regulated by the metastasis suppressor RhoGDI2 and is a novel promoter of tumor formation, lung metastasis and cancer cachexia. Oncogene 2006;26:765-73.
- Moriyama M, Sato T, Inoue H, Fukuyama S, Teranishi H, Kojima M, et al. The neuropeptide neuromedin U promotes inflammation by direct activation of mast cells. J Exp Med 2005;202:217-24.
- Fujii R, Hosoya M, Fukusumi S, Kawamata Y, Habata Y, Hinuma S, et al. Identification of neuromedin U as the cognate ligand of the orphan G protein-coupled receptor FM-3. J Biol Chem 2000;275:21068-74.
- Hosoya M, Moriya T, Kawamata Y, Ohkubo S, Fujii R, Matsui H, et al. Identification and functional characterization of a novel subtype of neuromedin U receptor. J Biol Chem 2000;275:29528-32.
- Kojima M, Haruno R, Nakazato M, Date Y, Murakami N, Hanada R, et al. Purification and identification of neuromedin U as an endogenous ligand for an orphan receptor GPR66 (FM3). Biochem Biophys Res Commun 2000;276:435-8.
- Shan L, Qiao X, Crona JH, Behan J, Wang S, Laz T, et al. Identification of a novel neuromedin U receptor subtype expressed in the central nervous system. J Biol Chem 2000;275:39482-6.
- Szekeres PG, Muir AI, Spinage LD, Miller JE, Butler SI, Smith A, et al. Neuromedin U is a potent agonist at the orphan G protein-coupled receptor FM3. J Biol Chem 2000;275:20247-50.
- Raddatz R, Wilson AE, Artymyshyn R, Bonini JA, Borowsky B, Adham N, et al. Identification and characterization of two neuromedin U receptors differentially expressed in peripheral tissues and the central nervous system. J Biol Chem 2000;275:32452-9.
- Qian J, Voorbach MJ, Huth JR, Coen ML, Zhang HC, Ng SC, et al. Discovery of novel inhibitors of Bcl-xL using multiple high-throughput screening platforms. Anal Biochem 2004;328:131-8.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099-108.
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67-73.
- Hashimoto T, Masui H, Uchida Y, Sakura N, Okimura K. Agonistic and antagonistic activities of neuromedin U-8 analogs substituted with glycine or D-amino acid on contractile activity of chicken crop smooth muscle preparations. Chem Pharm Bull (Tokyo) 1991;39:2319-22.
- Sakura N, Ohta S, Uchida Y, Kurosawa K, Okimura K, Hashimoto T. Structure-activity relationships of rat neuromedin U for smooth muscle contraction. Chem Pharm Bull (Tokyo) 1991;39:2016-20.
- Sakura N, Kurosawa K, Hashimoto T. Structure-activity relationships of neuromedin U. I. Contractile activity of dog neuromedin U-related peptides on isolated chicken crop smooth muscle. Chem Pharm Bull (Tokyo) 1995;43:1148-53.
- Hashimoto T, Kurosawa K, Sakura N. Structure-activity relationships of neuromedin U. II. Highly potent analogs substituted or modified at the N-terminus of neuromedin U-8. Chem Pharm Bull (Tokyo) 1995;43:1154-7.
- Kurosawa K, Sakura N, Hashimoto T. Structure-activity relationships of neuromedin U. III. Contribution of two phenylalanine residues in dog neuromedin U-8 to the contractile activity. Chem Pharm Bull (Tokyo) 1995;44:1180-4.
- Sakura N, Kurosawa K, Hashimoto T. Structure-activity relationships of neuromedin U. IV. Absolute requirement of the arginine residue at position 7 of dog neuromedin U-8 for contractile activity. Chem Pharm Bull (Tokyo) 2000;48:1166-70.
- Abiko T, Takamura Y. Synthesis of two neuromedin U (NMU) analogues and their comparative effect of reducing food intake in rats. Prep Biochem Biotechnol 2002;32:79-86.
- Wu B, Gao J, Wang MW. Development of a complex scintillation proximity assay for high-throughput screening of PPARγ modulators. Acta Pharmacol Sin 2005;26:339-44.
- Hui X, Gao J, Xie X, Suto N, Ogiku T, Wang MW. A robust homogeneous binding assay for α4β2 nicotinic acetylcholine receptor. Acta Pharmacol Sin 2005;26:1175-80.
- Go bel J, Saussy DL, Goetz AS. Development of scintillation-proximity assays for alpha adrenoceptors. J Pharmacol Toxicol Methods 1999;42:237-44.
- Gleich A, Schmidtchen FP, Mikulcik P, Müller G. Enantiodifferentiation of carboxylates by chiral building blocks for abiotic anion receptors. J Chem Soc Chem Commun 1990.55-7.
- Foye WO, Lemke TL, Williams DA (eds). Foye’s principles of medicinal chemistry; v 4. Pennsylvania: Williams and Wilkins; 1995. Chapters 11–14, 17, 18.
- Kajino M, Hinuma S, Tarui N, Yamashita T, Nakayama Y, inventors; Receptor regulator. EP 1 559 428 A1, 03.08.2005.