
Antiproliferative effect of rapamycin on human T-cell leukemia cell line Jurkat by cell cycle arrest and telomerase inhibition1
Introduction
The mammalian target of rapamycin (mTOR) pathway plays an important role in energy metabolism, the regulation of cell proliferation, and promoting cancer cell survival[1,2]. The critical functions of mTOR have led to the development of mTOR inhibitors (MTI), including rapamycin and its analogs as novel anticancer agents. The anticancer effects of MTI are presently evaluated in phase 1/2 clinical trials in various solid tumors. Moreover, the use of MTI is increasingly documented in hematological malignancies, such as chronic myeloid leukemia (CML), myelodysplastic syndromes (MDS), and acute myelogenous leukemia (AML)[3–6]. However, the antileukemic mechanism of rapamycin has not been extensively evaluated in acute T lymphoblastic leukemia (ATLL).
Telomerase is a ribonucleoprotein enzyme, which is necessary to compensate for telomere shortening during cell division by synthesizing telomere DNA. In most normal somatic cell types, telomerase is usually undetectable; however, the activation of telomerase is seen in cancer cells and is thought to be a critical element in cancer pathogenesis. Considering that telomerase activities were documented in some types of leukemia[7], and in addition, high telomerase activity and shortened telomere length were associated with poorer prognosis in adult T-cell leukemia[8], antitelomerase strategies in leukemia might be of considerable interest. As reported in an in vitro reconstitution assay, the human telomerase reverse transcriptase (hTERT), the rate-limiting factor for telomerase activity, could be phosphorylated at serine/threonine residues by protein kinase C and recombinant protein kinase B/Akt[9], but it remains unknown whether mTOR, a downstream of Akt, also affects telomerase activity. Thus, we used Jurkat cells, a cell line established from acute T lymphoblastic leukemia patient, to investigate the relationship between AKT/mTOR signaling and telomerase activity, and to explore the antileukemic mechanism of rapamycin in Jurkat cells.
In this work, we found that rapamycin displayed potent, antiproliferative activity against Jurkat cells, probably by inducing G1 phase arrest other than apoptosis. Moreover, exposure to rapamycin could reduce telomerase activity in Jurkat cells via the mTOR-mediated pathway, suggesting that rapamycin might be a novel potential treatment agent against acute T lymphoblastic leukemia.
Materials and methods
Cells culture and reagents Human T cell leukemia Jurkat cells were obtained from the Shanghai Institute for Biological Sciences, Chinese Academy of Science (Shanghai, China). The Jurkat cells were routinely cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco, USA) at 37 °C in a humidified atmosphere with 5% CO2. Exponentially-growing cells in a suspension were used in the experiments. Rapamycin and LY294002 were purchased from Sigma (St Louis, MO, USA), dissolved in Me2SO, and stored frozen at –20 °C. The antibodies against cyclin D1, cyclin D2, cyclin D3, cyclin-dependent kinase (CDK)4, CDK6, p27Kip1, p21waf1 and phospho-Akt (Ser473), phospho-p70S6K (Thr389), phospho-S6 (Ser235/236), as well as their non-phosphorylated antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Mouse anti-hTERT 2C4 and peroxidase-conjugated goat antimouse immunoglobulin M (IgM) were purchased from Abcam (Cambridge, MA, USA). Rabbit anti-actin antibody, goat antirabbit or antimouse immunoglobulin G (IgG) conjugated with horseradish peroxidase were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Assays of cell growth and apoptosis Cell growth was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric reduction method as previously described. Measurements were taken in quadruple at 24, 48, and 72 h after drug incubation at the indicated concentrations. Absorbance at 570 nm was measured in a BIO-RAD microplate reader (Hercules, California, USA). Apoptosis was measured by a flow cytometric analysis of cells stained with Annexin V-fluorescein-isothiocyanate (FITC) and propidium iodide (PI; BD PharMingen, San Diego, CA, USA) according to the manufacturer’s instructions. After incubation in Me2SO alone or rapamycin at the indicated concentrations for 72 h, the cells were pelleted by centrifugation and incubated with Annexin V-FITC and PI. Single-cell suspensions were analyzed by FACScan (Becton Dickinson, San Jose, CA, USA). Early apoptotic cells were scored as Annexin V+, PI-, whereas late apoptotic cells scored as Annexin V+, PI+ to exclude necrotic cells (Annexin V-, PI+).
Cell cycle analysis For the cell cycle analysis, 2×105 Jurkat cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), fixed in 70% ethanol, and incubated at 4 ºC overnight. The cells were then resuspended in 1 mL PBS containing 50 μg/mL PI and 100 U/mL RNAseA and incubated for 30 min at room temperature. The DNA content was monitored by flow cytometry. Cell cycle was analyzed with CellQuest software (Becton Dickinson, USA).
Western blot analysis The protein lysates were prepared from the control and drug-treated cells and separated by electrophoresis on 12% SDS-PAGE. The proteins were then transferred to polyvinylidene difluoride membranes. Subsequently, they were probed for molecules of interest with specific primary antibodies and then with goat antirabbit or antimouse IgG or IgM conjugated with horseradish peroxidase. The protein bands were visualized by a Western blot chemiluminescence reagent (KPL, Gaithersburg, MD, USA) according to the instructions of the manufacturer. For reprobing, the membranes were stripped (0.2 mmol/L NaOH) and reprobed with the desired antibodies. The molecular sizes of the protein bands were determined by comparison with prestained protein markers (M-0671; Sigma, USA).
Telomeric repeat amplication protocol assay A PCR-ELISA-based assay (Roche Molecular Biochemicals, Mannheim, Germany) was used to measure the telomerase activity, as described in detail by the manufacturer. In total, 2×105 cells were lysed with 200 μL lysis buffer on ice for 30 min. After the pelleting of cellular debris by centrifugation, the supernatant was stored at -80 °C until further use. Each telomeric repeat amplication protocol (TRAP) reaction contained 1 mg of total protein. The telomerase substrate primer was elongated by telomerase at 25 ºC for 30 min, and then incubated at 94 ºC for 5 min to induce the telomerase inactivation. The reaction mixture was then amplified by 30 cycles of PCR at 94 ºC for 30 s, 50 ºC for 30 s, 72 ºC for 90 s, and 72 ºC for 10 min. The 5 μL PCR products were denatured, hybridized to a digoxigenin-labeled probe, and detected by ELISA. Relative telomerase activity (RTA) was calculated as follows: RTA=(ODexp-ODneg)/(ODpos-ODneg)×100%. As a positive control, the human epithelial carcinoma cell line HeLa was used; as a negative control, a positive sample that had been subjected to heating (85 °C) for 20 min prior to the TRAP assay was used.
RT-PCR analysis Total cellular RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and isolated with the single-step acid guanidinium-isothiocyanate/phenol-chloroform extraction method. In total, 1 μL total RNA (approximately 1 ng) was used to synthesize cDNA with random primers and Moloney murine leukemia virus RT (Promega, Madison, WI, USA). hTERT mRNA (619 bp) was amplified with the following primers: sense, 5´-CGGAAGAGTGTCTGGAGCAA-3´; antisense, 5´-GGATGAAGCGGAGTCTGGA-3´. β-Actin mRNA (144 bp) was amplified with the following primers: sense, 5´-CGCTGCGCTGGTCGTCGACA-3´; antisense, 5´-GTCACGCACGATTTCCCGCT-3´. After an initial denaturation for 3 min at 94 °C, cDNA was amplified in a final volume of 25 μL with1.25 units Taq DNA polymerase (Promega, USA). The amplification consisted of 36 cycles (hTERT) or 28 cycles (β-actin) for 45 s at 94°C, 60 s at 60°C, and 60 s at 72°C. The PCR product (10 μL) was subjected to electrophoresis on 1.5% agarose containing ethidium bromide and visualized by UV absorption. The gels were photographed, and the bands were analyzed by computerized densitometry. β-Actin mRNA was used as the internal control.
Statistical analysis The data were presented as mean±SD and analyzed by SPSS 11.0 software (SPSS, Chicago, IL, USA) using Student’s t-test and ANOVA. Differences were considered statistically significant at P<0.05.
Results
Effect of rapamycin on growth, cell cycle, and apoptosis in Jurkat cells Exposure to rapamycin (0.1–100 nmol/L) resulted in a significant inhibitory effect on the proliferative activity of Jurkat cells: an effect that was dose- and time-dependent (Figure 1A). The 50% inhibitory concentrations (IC50) values for 24, 48, and 72 h were 344, 92, and 16 nmol/L, respectively. To determine the mechanism of growth inhibition by rapamycin in more detail, we analyzed the cell cycle profile after treatment with rapamycin. As expected, G1 phase arrest was induced by rapamycin (100 nmol/L) after 16 h, and the ratio of G1 continued to grow as treatment was prolonged to 32 h (Figure 1B). Furthermore, we evaluated the effect of rapamycin on the cell cycles of synchronized cells by serum starvation. Serum starvation resulted in the accumulation of cells in the G1 phase. Serum stimulation resulted in the transition of cells from the G1 phase to the S phase with a concomitant decrease in the G1 phase. Rapamycin significantly blocked serum-induced entry to the S phase in a dose-dependent manner in Jurkat cells (Figure 1C). In addition, rapamycin did not increase the amount of cells in the Annexin V+ fraction 72 h after treatment, confirming no increase in apoptosis (Figure 1D). These results suggest that rapamycin inhibits cell growth via cell cycle arrest and not by the induction of apoptosis in Jurkat cells.
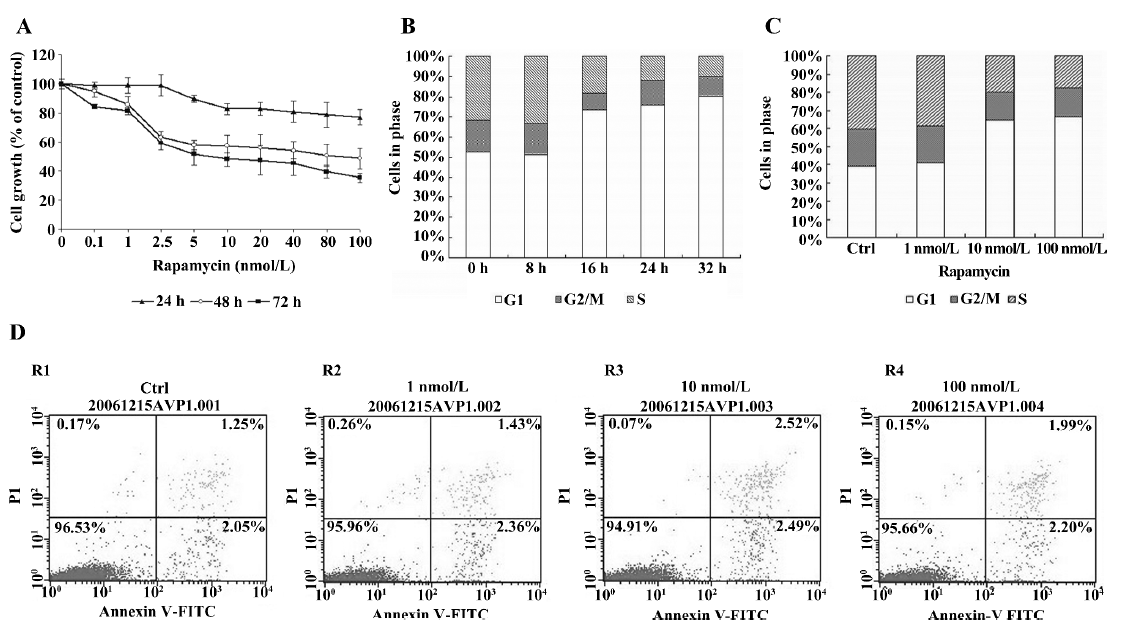
Expression of p27Kip1, p21waf1, CDK4, CDK6, cyclin D1, cyclin D2, and cyclin D3 in Jurkat cells treated with rapamycin We investigated the effects of rapamycin on G1 phase proteins, specifically p27Kip1, p21waf1, CDK4, CDK6, cyclin D1, cyclin D2, and cyclin D3 (Figure 2). The Jurkat cells were serum deprived for 16 h, and then stimulated with 10% serum and either rapamycin or ME2SO as the control for 12, 24, and 48 h. First, the levels of p27Kip1 and p21waf1 were high in serum-starved Jurkat cells (0 h), but underwent rapid elimination upon the addition of serum. Rapamycin treatment maintained elevated levels of p27Kip1 and p21waf1. Cyclin D1 was not detected in the Jurkat cells under any conditions tested, which is in agreement with previously published observations[10]. Rapamycin did not affect the protein levels of either CDK4 or CDK6; however, rapamycin downregulated cyclin D3 without affecting the cyclin D2 protein levels. This decrease in cyclin D3 protein levels could be observed as early as 12 h after release from serum starvation, and it persisted throughout the 48 h experiment period.
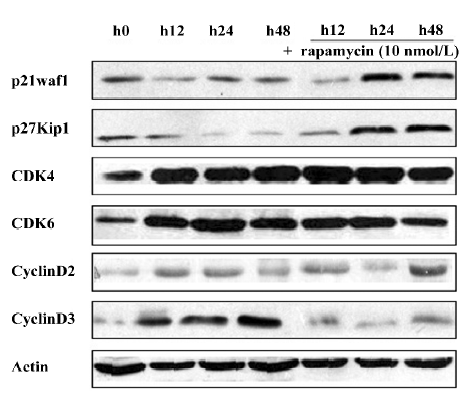
Effect of rapamycin on the PI3K/Akt/mTOR signal transduction pathway Much is already known about the PI3K/Akt/mTOR signaling pathway and its modulation of cell growth. To ascertain it, we characterized the effect of rapamycin on the cell signaling pathways on Jurkat cells. The cells were treated with 10 nmol/L rapamycin or 50 μmol/L LY294002 for 0–24 h. As a PI3K inhibitor, LY294002 inhibited Akt phosphorylation at Ser473. Inconsistent with a previous model that rapamycin functioned downstream of Akt[11], rapamycin treatment was found to slightly downregulate the phosphorylation of Akt (Figure 3A). Previous studies suggested that p70S6K is a downstream target of mTOR[12]. The p70S6K kinase directly phosphorylates the 40S ribosomal protein S6, which results in the enhanced translation of proteins that contain a polypyrimidine tract in the 5´-untranslated region[13]. Therefore, the effect of rapamycin on the phosphorylation of p70S6K and S6 was examined. As a result, rapamycin markedly reduced the phosphorylation of p70S6K and S6. This effect was seen as early as 12 h after exposure to rapamycin, and the total proteins of the pan-S6 and p70S6K maintained stable levels (Figure 3B). These experimental data suggest that rapamycin exerts its effect through the PI3K/Akt/mTOR/p70S6K pathway by regulating the phosphorylation of p70S6K and S6 proteins.
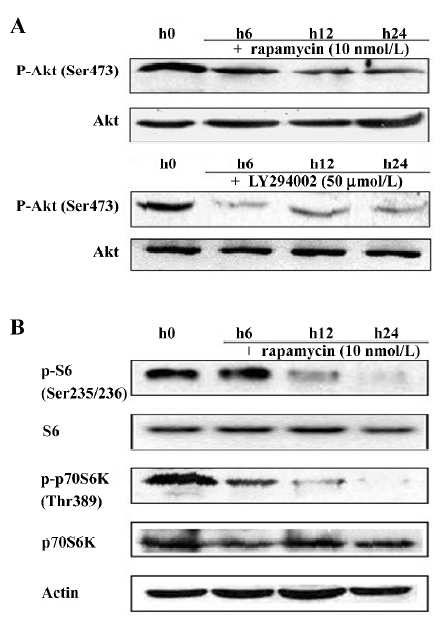
Rapamycin inhibits telomerase activity in Jurkat cells To date, it remains unclear how telomerase activity is differentially regulated by mTOR signaling in various cell systems, including the human T-cell leukemia cell line. Thus, we were interested in the regulation of telomerase by rapamycin in Jurkat cells. First, telomerase activity was measured by the TRAP assay. When the cells were treated with rapamycin, telomerase activity was downregulated within 24 h and had a reduced tendency that was dose- and time-dependent (Figure 4A). Because telomerase activity correlates with the expression of hTERT mRNA[14], we used RT-PCR to examine the expression of hTERT mRNA in Jurkat cells treated with either rapamycin or Me2SO as the control for 48 h. We found that treatment with varying concentrations of rapamycin significantly reduced the hTERT mRNA level in a dose-dependent manner (Figure 4B). Furthermore, by Western blotting with the 2C4 monoclonal antibody specific for hTERT, we noted that the hTERT protein level decreased and correlated with these changes in the expression of hTERT mRNA induced by rapamycin (Figure 4C). These data suggest that rapamycin may inhibit telomerase activity by rapidly decreasing the hTERT mRNA levels.
Discussion
Acute lymphoblastic leukemia (ALL) is the most common type of leukemia in young children. This disease also affects adults, especially those aged 65 years or older. While the outcome for children with ALL has improved dramatically with current therapy, children with relapsed ALL and adults with ALL often develop resistance to standard chemotherapy. Even under aggressive therapy, these patient groups have 5 year disease-free survival rates of only 28%–39%[15]. Evidence accumulated over recent years has indicated the phosphoinositide 3-kinase/Akt/mTOR signal transduction pathways as one of the major factors implicated in human leukemia resistance to conventional chemotherapy[16]. Thus, the development of novel therapeutic agents override the resistance, and directly targeting this signaling network is crucial. One potential class of novel therapeutics is MTI, such as rapamycin.
Rapamycin, a macrocyclic lactone produced by Streptomyces hygroscopicus, was the first MTI to be used clinically. Initially developed as an immunosuppressive agent, rapamycin is well tolerated in humans. Preclinical studies on human cancer cell lines and human tumor xenograft models have shown that rapamycin may be effective for the treatment of prostate, small cell lung, glioblastoma, renal cell, and breast cancer[17]. Moreover, the use of MTI is increasingly documented in hematological malignancies, such as CML, MDS, and AML[3–6]. To data, however, the antileukemic mechanism of rapamycin has not been extensively evaluated in ATLL. In this work, we used the human T-cell leukemia cell line Jurkat to study the antileukemic effect of rapamycin.
First, we observed that rapamycin could greatly inhibit the proliferation of Jurkat cells in dose- and time-dependent manners in vitro. To further determine the mechanism of growth inhibition by rapamycin, we performed an apoptosis assay with flow cytometry. As previously demonstrated in other cancer cell lines, rapamycin did not significantly induce apoptosis in the Jurkat cells, indicating that it may exert antiproliferative effects via other mechanisms. It is now well established that besides apoptosis, cell cycle regulation in the G1 phase may be a promising target for the development of new chemotherapeutic anticancer agents. Various molecules, including cyclins, CDK, and CDK inhibitors play important roles in controlling major checkpoints in the cell cycle. Their alterations could lead to tumorgenesis. Cyclin D acts as a sensor of extracellular mitogenic signals and plays a critical, rate-limiting role in cell cycle progression during mid G1 by initiating the multistep process that leads to pRb inactivation[18,19]. p27Kip1 and p21waf1 act as a negative regulator of cyclins and CDK activity, and appear to be essential to arrest cells before the late G1 restriction point[20,21]. We performed a cell cycle analysis in our work and found that rapamycin significantly induced G1 arrest and blocked serum-induced entry to the S phase in a dose-dependent manner in Jurkat cells. Rapamycin downregulated cyclin D3 without affecting the cyclin D2, CDK4 or CDK6 protein levels, and maintained elevated levels of p27Kip1and p21waf1. Therefore, we concluded that rapamycin potently suppressed proliferation in Jurkat cells partly via the induction of G1 cell cycle arrest, which was mediated by affecting cyclin D3, p27Kip1,and p21waf1. To understand the mechanisms by which rapamycin functions, we characterized the signaling pathways affected by rapamycin. A previous model showed that rapamycin functioned downstream of Akt[11]; unexpectedly, we found that rapamycin treatment also slightly downregulated the phosphorylation of Akt. This could be explained by data obtained by others showing that rapamycin inhibits the assembly of mTORC2, which phosphorylates and activates Akt[22]. Furthermore, we found that rapamycin reduced the phosphorylation of mTOR downstream cascade targets p70S6K and S6 in Jurkat cells. As it is known, p70S6K modulates the initiation of translation and cell cycle progression through downstream effectors, such as cyclin D, CDK4 and CDK6[23], so it is possible to assert that rapamycin inhibits G1 cell cycle progression through affecting the mTOR/p70S6K-signaling pathway.
The major novel finding from this work is that rapamycin potently suppresses telomerase activity in Jurkat cells. Information about the regulation of telomerase by rapamycin is presently incomplete, even if some data have been documented in endometrial cancer cells[24]. Telomerase, the ribonucleoprotein complex involved in telomere maintenance, is composed of 2 main components: the telomerase RNA template[25] and hTERT[26]. In most normal somatic cell types, telomerase activity is usually repressed[27]. The activation of telomerase is seen in cancer cells, including leukemia cells, and is thought to be a critical element in oncology pathogenesis[28]. Thus, telomerase expression and activity can serve as a molecular marker of the clinical progression and prognosis of most leukemias. Antitelomerase strategies have been proven to be promising in some types of leukemias[7]. With regards to hTERT, the rate-limiting factor for telomerase activity[29], we determined the hTERT mRNA and protein levels in Jurkat cells treated with rapamycin and found that rapamycin exposure resulted in significantly decreased levels of hTERT mRNA and protein. This implies that rapamycin may exert its effect on telomerase through the regulation of hTERT gene transcription. In mammalian cells, the expression of various transcription factors, such as E2F, hypoxia-inducible factor 1a (HIF-1a), and signal transducer and activator of transcription 3 (STAT3) seems to be modulated by the mammalian target of rapamycin[30–32]. Hirotaka et al reported that the hTERT promoter region between –165 and +51 contains 2 HIF-1 consensus motifs[33]. However, the exact transcription factors, which modulate hTERT gene transcription and are targeted by the mTOR pathway, need to be further identified.
Although the exact mechanisms are not clear, there may be some alternative pathways involved in telomerase downregulation by rapamycin, other than the direct regulation of hTERT gene transcription. A potential connection between rapamycin and telomerase regulation is through rapamycin’s effect on the cell cycle. Zhu et al reported that telomerase activity was high during the S phase, but undetectable in the G2-M phase[34]. Our group also found that the expression of hTERT and telomerase activity were both upregulated in normal mesenchymal stem cells during their transit through the S phase (data not shown). If hTERT transcription is cell cycle dependent, then the effect of rapamycin on hTERT expression could be the indirect consequence of cell cycle arrest. As earlier mentioned, p27Kip is essential in blocking the G1-S transition. Recent studies have shown that the overexpression of p27 suppresses hTERT promoter activity and telomerase activity in mouse embryonic fibroblasts (MEF) deficient in p27 (p27-/- MEF)[35], which implies that hTERT transcription is cell cycle dependent. We also found that rapamycin upregulates the p27 protein level in Jurkat cells, which might correlate with the downregulated hTERT expression. In addition, as reported in an in vitro reconstitution assay, hTERT was an Akt kinase substrate protein, and human telomerase activity was enhanced through the phosphorylation of the hTERT protein by recombinant protein kinase B/Akt[9]. In our work, we demonstrated that rapamycin treatment reduced the phosphorylation of Akt. Therefore, rapamycin might downregulate hTERT peptide phosphorylation and telomerase activity together through suppressing the phosphorylation of Akt.
Taken together, our findings suggest that rapamycin displays potent antileukemic effects in the human T-cell leukemia cell line by the inhibition of cell proliferation through G1 cell cycle arrest and also through the suppression of telomerase activity. Since telomerase activity appears to play a critical role in leukemiagenesis, the use of a mTOR inhibitor, such as rapamycin, is a promising targeted therapeutic strategy and may have significant clinical implications in the treatment of some leukemias.
Acknowledgements
We thank Dr Jian-ping LAN from Zhejiang Provincial People’s Hospital for insightful discussion during the course of this study. We also gratefully acknowledge the generous help from members of the Institute of Hematology at The First Affiliated Hospital of Zhejiang University Medical School.
References
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science 2001;294:1102-5.
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 2000;103:253-62.
- Panwalkar A, Verstovsek S, Giles FJ. Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer 2004;100:657-66.
- Follo MY, Mongiorgi S, Bosi C, Cappellini A, Finelli C, Chiarini F. The Akt/mammalian target of rapamycin signal transduction pathway is activated in high-risk myelodysplastic syndromes and influences cell survival and proliferation. Cancer Res 2007;67:4287-94.
- Mayerhofer M, Aichberger KJ, Florian S, Krauth MT, Hauswirth AW, Derdak S, et al. Identification of mTOR as a novel bifunctional target in chronic myeloid leukemia: dissection of growth-inhibitory and VEGF-suppressive effects of rapamycin in leukemic cells. FASEB J 2005;19:960-2.
- Martelli AM, Tazzari PL, Evangelisti C, Chiarini F, Blalock WL, Billi AM, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedside. Curr Med Chem 2007;14:2009-23.
- Deville L, Hillion J, Lanotte M, Rousselot P, Segal Bendirdjian E. Diagnostics, prognostic and therapeutic exploitation of telomeres and telomerase in leukemias. Curr Pharm Biotechnol 2006;7:171-83.
- Kubuki Y, Suzuki M, Sasaki H, Toyama T, Yamashita K, Maeda K, et al. Telomerase activity and telomere length as prognostic factors of adult T-cell leukemia. Leuk Lymphoma 2005;46:393-9.
- Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 1999;274:13085.
- Lei W, Liu F, Ness SA. Positive and negative regulation of c-Myb by cyclin D1, cyclin-dependent kinases, and p27 Kip1. Blood 2005;105:3855-61.
- Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 2000;60:3504-13.
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 1995;377:441-6.
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5’TOP mRNA translation through inhibition of p70s6k. EMBO J 1997;16:3693-704.
- Kyo S, Kanaya T, Takakura M, Tanaka M, Inoue M. Human telomerase reverse transcriptase as a critical determinant of telomerase activity in normal and malignant endometrial tissues. Int J Cancer 1999;80:60-3.
- Gokbuget N, Hoelzer D. Recent approaches in acute lymphoblastic leukemia in adults. Rev Clin Exp Hematol 2002;6:114-41.
- West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resistance Updates 2002;5:234-48.
- Rowinsky EK. Targeting the molecular target of rapamycin (mTOR). Curr Opin Oncol 2004;16:564-75.
- Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993;7:817-21.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323-30.
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of P27kip1, a cyclin-dependent kinase inhibitor and potential mediator of extracellular antimitogenic signals. Cell 1994;78:59-66.
- Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994;78:67-74.
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006;22:159-68.
- Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene 2000;19:6680-6.
- Bae-Jump VL, Zhou C, Gehrig PA, Whang YE, Boggess JF. Rapamycin inhibits hTERT telomerase mRNA expression, independent of cell cycle arrest. Gynecol Oncol 2006;100:487-94.
- Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, et al. The RNA component of human telomerase. Science 1995;269:1236-41.
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997;277:955-9.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349-52.
- Stewart SA, Weinberg RA. Telomerase and human tumorigenesis. Semin Cancer Biol 2000;10:399-406.
- Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev 2002;66:407-25.
- Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22:7004-14.
- Bruemmer D, Yin F, Liu J, Kiyono T, Fleck E, Van Herle AJ, et al. Rapamycin inhibits E2F-dependent expression of minichromosome maintenance proteins in vascular smooth muscle cells. Biochem Biophys Res Commun 2003;303:251-8.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004;18:1926-45.
- Nishi H, Nakada T, Kyo S, Inoue M, Shay JW, Isaka K. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol 2004;24:6076-83.
- Zhu X, Kumar R, Mandal M, Sharma N, Sharma HW, Dhingra U, et al. Cell cycle-dependent modulation of telomerase activity in tumor cells. Proc Natl Acad Sci USA 1996;93:6091-5.
- Lee SH, Kim JW, Oh SH, Kim YJ, Rho SB, Park K, et al. IFN-gamma/IRF-1-induced p27kip1 down-regulates telomerase activity and human telomerase reverse transcriptase expression in human cervical cancer. FEBS Lett 2005;579:1027-33.