
Levistolide A overcomes P-glycoprotein-mediated drug resistance in human breast carcinoma cells1
Introduction
The multidrug resistance (MDR) phenotype is often associated with the overexpression of ATP-binding cassette (ABC) transporters, which can actively extrude a wide variety of structurally diverse anticancer agents, thereby reducing their intracellular drug concentrations[1]. P-glycoprotein (P-gp) is a 170 kDa transmembrane glycoprotein encoded by the MDR1 gene[2,3]. The overexpression of P-gp was shown to be resistant against various clinically relevant compounds, including anthracyclines and vinca alkaloids[1]. In addition, several cytological dyes, such as 3-ethyl-2-(3-[3-ethyl-2{3H}-benzoxazolylidene]-1-propenyl) benzoxazolium iodide [DiOC2(3)] demonstrated decreased accumulation in P-gp overexpressing cells[4]. P-gp overexpression appears to be closely correlated with poor prognosis for cancers, including breast carcinoma[5]. Therefore, a successful inhibition of the P-gp transporter function or its expression may overcome MDR by increasing the intracellular accumulation of anticancer drugs.
Although many agents have been identified as competitive antagonists of P-gp in vitro, most have little or no therapeutic potential because of high toxicity in vivo at the doses required to reverse MDR. For instance, verapamil (VER), a calcium channel blocker used in the treatment of ischemic heart disease, is a highly effective competitive antagonist of P-gp, but the doses required to overcome MDR result in unacceptable cardiovascular toxicity[6].
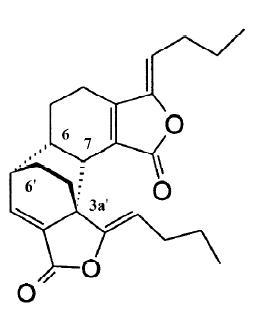
Levistolide A (LA) is a naturally occurring compound extracted from the rhizomes of Angelicae sinensis (Oliv). In Chinese traditional medicine, the root of Angelicae sinensis is used for tonifying circulation and removing blood stasis. It has been applied in clinics for treating gynecological symptoms, anemia, chronic bronchitis, asthma, rheuma-tism, and other diseases[7]. It is also used as an essential component of antitumor formulas in China. The most important effect of Angelicae sinensis in antitumor formulas is to enhance the efficacy and reduce the toxicity of chemotherapy[8–10]. Therefore, we hypothesized that some of the components in Angelicae sinensis may serve to enhance the efficacy of chemotherapy. We screened more than 50 fractions in the organic solution and found that LA had a potent capacity to sensitize MDR cells to a variety of chemotherapy drugs, including adriamycin (ADR) and vincristine (VCR). In this study, we investigated the reversing effect of LA on P-gp overexpressing Bcap37/MDR1 cells and compared its reversing efficiency with a typical P-gp modulator VER.
Materials and methods
Cell culture and reagents Bcap37 breast carcinomas cells and P-gp overexpressing subline Bcap37/MDR1 cells derived by transfecting the human MDR1 gene[11], were generously provided by Dr Cao JIANG at Zhejiang University (Hangzhou, China). These cell lines were cultured in RPMI-1640 medium supplemented with 10% fatal bovine serum (FBS) in 5% CO2 at 37 ºC. LA used in this study was purified from Angelicae sinensis according to the reported methods[12]. Its structural identity was determined spectroscopically (1H and 13C NMR, and MS) as described previously[13,14]. ADR, VCR, VER, DiOC2(3), and dimethyl thiazolyl-2,5-diphenyltetrazolium bromide (MTT) were from Sigma (St Louis, MO, USA).
MTT assay Chemosensitivity was measured by the MTT assay. Briefly, the cells were seeded into 96-well plates and allowed to attach for 24 h, then the drugs diluted with the medium at different concentrations were added into the wells. To determine the reversal effect, graded concentrations of ADR or VCR with or without P-gp modulators were added. Then the cells were exposed to the drugs for 68 h at 37 °C, after which 20 µL MTT (5 mg/mL diluted in phosphate-buffered saline [PBS]) was added to each well and incubated for an additional 4 h. The formed formazan was dissolved in DMSO after aspirating the culture medium. The plates were shaken mechanically and the optical density of each well was read on a microplate reader (Opsys MR, Dynex Technology, Chantilly, Virginia, USA) at 470 nm. The reversal activity of VER or LA on ADR and VCR resistance was expressed as the reversing factors (RF), which were calculated according to the following equation: RF=IC50 antitumor drug alone/IC50 antitumor drug+modulator.
Cell cycle analysis The cells were seeded into 6-well plates and allowed to attach for 24 h. Then various concentrations of drugs diluted with the medium were added into the wells and incubated for 24 h. After the treatment period, the cells were harvested and fixed in 70% ethanol. The cells were again washed with PBS and then incubated with propidium iodide (10 µg/mL) with simultaneous treatment of RNase at 37 °C for 30 min. The percentages of the cells in the different phases of the cell cycle were measured with a FACStar flow cytometer (Becton Dickinson, San Jose, CA, USA) and analyzed using ModFit software (Verity, Topsham, USA).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling assay The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay was performed using the commercial kit (Roche Molecular Biochemicals, Indianapolis, USA) according to the manufacturer’s instructions[15]. Briefly, the cells were collected and fixed in 1% formaldehyde in PBS for 15 min on ice, washed with PBS, resuspended in 70% ethanol, and stored overnight at -20 °C. After rehydration in PBS for 5 min, the cells were resuspended in 50 µL reaction buffer (5 U terminal transferase, 2.5 mmol/L cobalt chloride, 0.5 mmol/L biotin-dUTP, and TdT buffer containing 0.2 mol/L potassium cacodylate, 25 mmol/L Tris–HCl, and 0.25 mg/mL bovine serum albumin, pH 6.6) and incubated at 37 °C for 30 min. The cells were then washed with PBS containing 0.1% Triton X-100, resuspended in 100 µL staining buffer, and incubated for 30 min in the dark. The pellets were then washed twice with rinsing buffer, resuspended in PBS, and measured with a flow cytometer.
Western blot analysis The cell extracts were equally loaded onto 8%–12% SDS–PAGE and electrophoretically transferred to NC membranes (Bio-Rad, New Orleans, Louisiana, USA). The membranes were blocked with 5% skimmed milk in PBS containing 0.1% Tween 20 (PBST) and incubated overnight with each primary antibody against P-gp (Chemicon Tech, Hangzhou, China), poly(ADP-ribose) polymerase (PARP; Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase-3 (Cell Signaling Tech, Danvers, MA, USA), and β-actin (Sigma, St Louis, USA). The membranes were then washed with PBST and incubated for 1 h with a secondary antibody and visualized with an enhanced chemiluminescence detection kit (Pierce, Appleton, WI, USA).
RNA extraction and RT–PCR The total RNA from Bcap37/MDR1 cells was extracted using TRIzol and isolated according to the procedure supplied by the manufacturer (Invitrogen, USA). Reverse transcription was carried out according to the manufacturer’s instructions (GIBCO-BRL, USA). The first strand of the cDNA was generated from 5 µg of total RNA using the oligo(dT) primer and superscript II reverse transcriptase (GIBCO-BRL, USA). The cDNA were amplified by PCR using the Supermix kit (Toyobo, Tokyo, Japan). The primer sets for P-gp were 5'-CCC ATC ATT GCA ATA GCA GG-3' and 5'-GTT CAA ACT TCT GCT CCT GA-3'; and the primer sets for β-actin were 5'-GTG GGG CGC CCC AGG CAC CA-3' and 5'-CTC CCT AAT GTC ACG CAC GAT TTC-3'. Amplified DNA fragments were electrophoresed on 1% agarose gels.
DiOC2(3) efflux assays The fluorescence intensity of intracellular DiOC2(3) was determined by flow cytometry. Briefly, the Bcap37/MDR1 cells were harvested, incubated with 0.1 µmol/L DiOC2(3) at 37 °C for 1 h, washed with ice-cold PBS, and resuspended in fresh medium containing P-gp modulators for indicated time. Then the cells were washed with ice-cold PBS and the mean fluorescence intensity of intracellular DiOC2(3) was detected using flow cytometry.
Statistical analysis All experiments were repeated at least 3 times and expressed as mean±SD. The statistical difference between the control and treatment groups was determined by t-test. A P-value of <0.05 was considered to be statistically significant.
Results
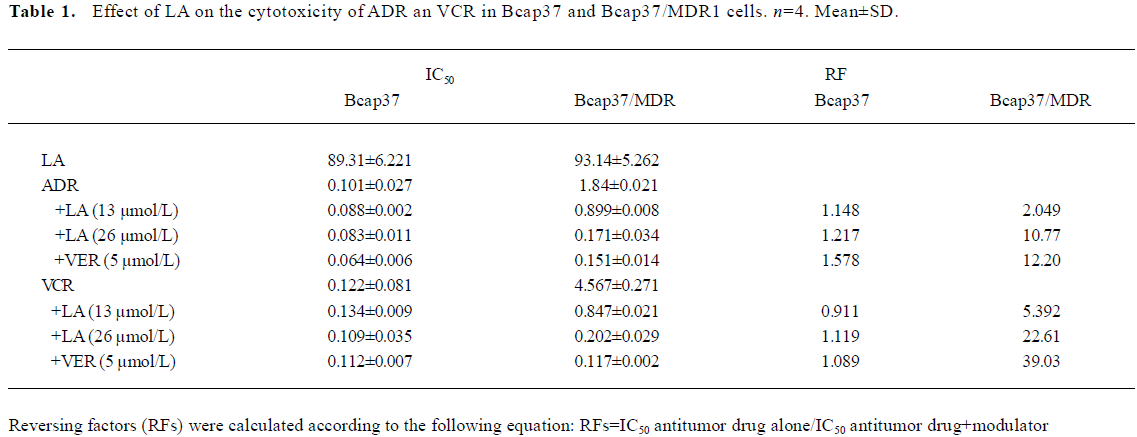
LA enhances ADR- or VCR-induced G2/M arrest in Bcap37/MDR1 cells The chemosensitivity of MDR cells to the chemotherapy drugs was measured by IC50 comparison expressed as RF. The IC50 of LA were 89.31 and 93.14 µmol/L, respectively, in Bcap37 and Bcap37/MDR1 cells (Table 1). At 39 µmol/L (15 µg/mL) or less, LA did not cause any cytotoxicity to Bcap37 or Bcap37/MDR1 cells (Figure 1). Therefore, 13, 26, and 39 µmol/L were chosen as the working concentrations in the next experiments. As expected, the Bcap37/MDR1 cells were more resistant to ADR and VCR. The IC50 for ADR and VCR without a modulator in the Bcap37 cells were 0.10 and 0.11 µmol/L, respectively (Table 1), while in the Bcap37/MDR1 cells, the IC50 for ADR and VCR without a modulator were 1.84 and 4.57 µmol/L, respectively (Table 1). LA dose dependently increased the sensitivity of Bcap37/MDR1 to ADR and VCR (Figure 2A). At a concentration of 26 µmol/L, LA shifted the IC50 for ADR and VCR to 0.17 and 0.2 µmol/L with RF of 10.70- and 22.46-fold, respectively (Table 1). However, LA treatment did not significantly affect drug sensitivity in the Bcap37 cells (Table 1).
Full table
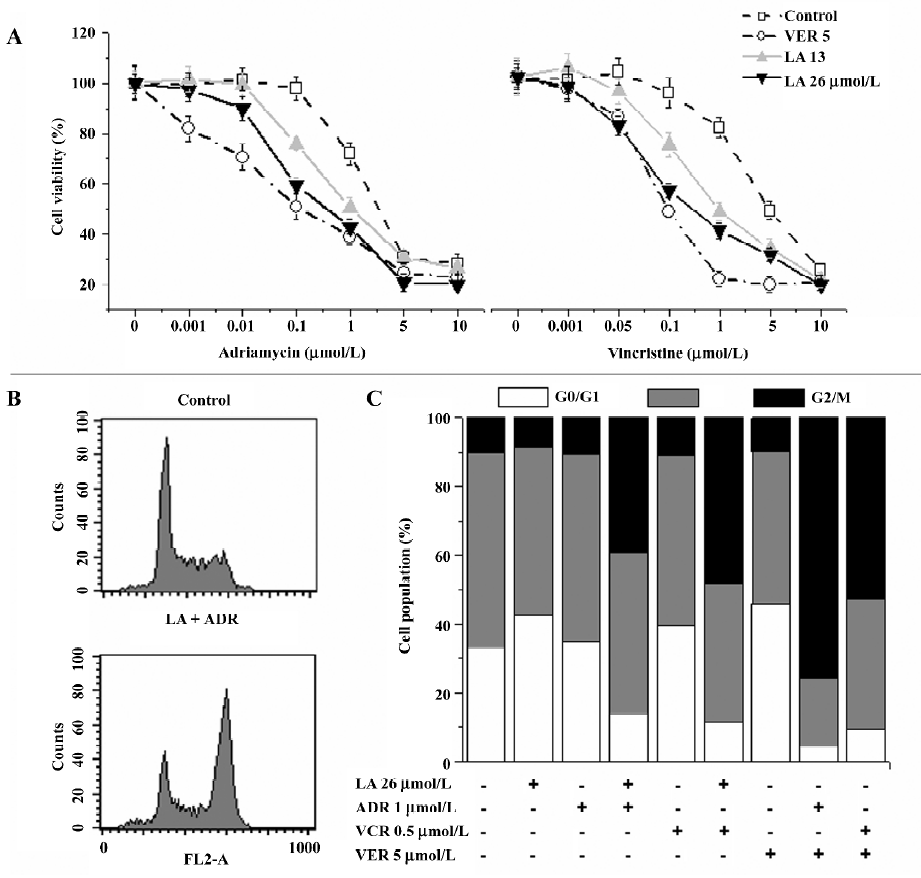
Usually, agents that induce cytotoxicity by damaging DNA (e.g. ADR) or inhibiting tubulin polymerization (e.g. VCR) arrest cell cycle at the G2/M phase[16]. As expected, we found ADR (2 µmol/L and above) and VCR (5 µmol/L and above) dose-dependently induced G2/M arrest (data not shown). Neither ADR (1 µmol/L) nor LA (26 µmol/L) alone caused cell cycle arrest, whereas treatment with a combination of both induced a significant G2/M arrest up to 60.94% (Figure 2B). However, higher doses of LA (>54 µmol/L) induced cell cycle arrest at the G0/G1 phase (data not shown), suggesting that LA enhanced the ADR-induced G2/M arrest. Similarly, we found that the VCR-induced G2/M arrest was also increased by LA by 82.04% (Figure 2B). The potentiated effects of LA (26 µmol/L) on G2/M arrest were a little less than those of VER at 5 µmol/L. These results suggest that like VER, LA enhanced ADR- or VCR-induced G2/M arrest, which may attribute its sensitizing effect to drug resistance.
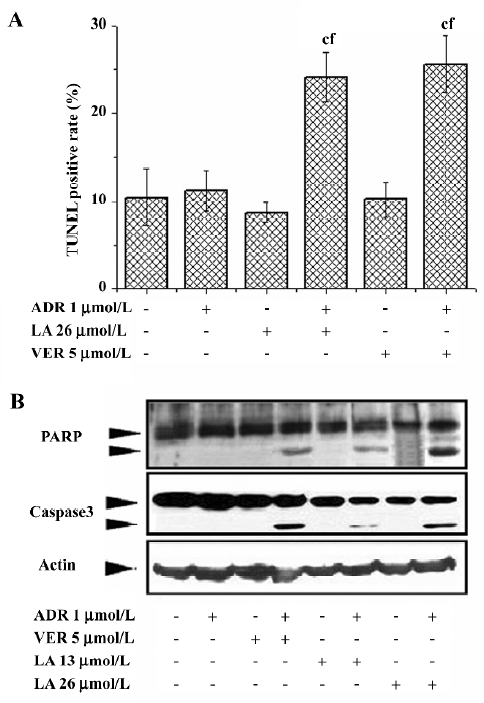
LA treatment potentiates ADR-induced apoptosis in Bcap37/MDR1 cells Inducing apoptosis is the main mechanism for ADR and VCR to treat carcinomas. We therefore investigated whether LA increased the cytotoxicity of ADR or VCR by apoptosis. Apoptosis was determined by TUNEL assay. Treatment with ADR (1 µmol/L), VER (5 µmol/L), or LA (26 µmol/L) alone did not increase TUNEL-positive cells compared to the control, while the cotreatment of ADR with LA or VER significantly increased TUNEL staining up to 53.6% and 59.8%, respectively (Figure 3A). Similarly, LA (26 µmol/L) cotreatment with VCR (0.5 µmol/L) significantly enhanced VCR-induced TUNEL staining (data not shown). The effect of LA (13 and 26 µmol/L) on apoptosis potentiation was further evidenced by caspase-3 activation and PARP cleavage. As illustrated in Figure 3B, caspase-3 was activated and PARP was cleaved in VER and ADR (or VCR)-cotreated MDR cells, but not in ADR (or VCR) alone-treated cells. Similar to VER, cotreatment with LA also caused a notable increase of caspase-3 activation and PARP cleavage (Figure 3B). These results suggest that LA treatment might inhibit P-gp function and subsequently lead to an increase in its apoptotic potential resulting from the intracellular accumulation of anticancer drugs.
LA inhibits DiOC2(3) efflux out of Bcap37/MDR1 cells without affecting P-gp expression In order to determine whether LA reversed MDR by inhibiting P-gp function, we investigated the effect of LA on the intracellular accumulation of a specific P-gp substrate DiOC2(3). As illustrated in Figure 4A, the inhibition of P-gp with LA dose dependently caused an increase in the accumulation of DiOC2(3) in MDR cells. LA at 39 µmol/L increased drug accumulation by 6.5-fold, with the efficiency similar to that of VER at 10 µmol/L (Figure 4A). P-gp is a drug transporter and functions by pumping its substrates out of MDR cells. By increasing the intracellular DiOC2(3) accumulation of LA suggests that LA has a capacity to suppress P-gp function. To determine whether LA also affects P-gp expression, we exposed the MDR cells to LA for indicated times, then detected MDR1 gene transcription by RT–PCR and P-gp protein expression by Western blotting. As shown in Figure 4B, LA at 26 µmol/L did not affect MDR1 mRNA or P-gp expression during 48 h exposure. These results indicate that LA overcame P-gp-mediated MDR by inhibiting P-gp function without affecting the P-gp expression.
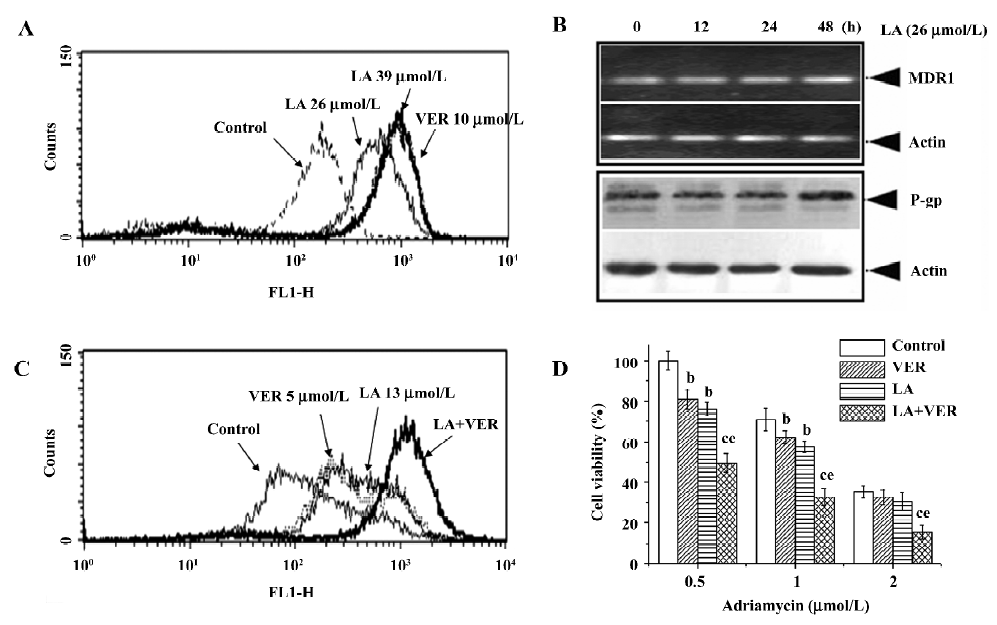
LA inhibits LA-enhanced VER ability to reverse drug resistance To investigate whether LA targets the same site of VER or other modulators, the MDR cells were incubated with paired modulators and then their effects on DiOC2(3) accumulation were analyzed. Cotreatment with LA (13 µmol/L) and VER (5 µmol/L) for 2 h dramatically increased DiOC2(3) accumulation much more than LA (13 µmol/L) or VER (5 µmol/L) alone (Figure 4C). After incubation for 2 h, the drug accumulation peaks became more flat than those of incubation at 1 h (Figure 4A) in the control, VER, or LA alone-treated groups, suggesting the drug efflux was over influx in these groups (Figure 4C). However, in the LA and VER cotreated group, the peak remained sharp (Figure 4C), suggesting that cotreatment significantly potentiated the efflux pump inhibitory effect of LA or VER. However, we did not show similar results in the pair of LA and cyclosporin A (CsA), or of VER and CsA (data not shown). It is therefore reasonable to hypothesize that by using relatively low concentrations of LA and VER, it may be possible to develop a less toxic regimen to reverse MDR. As illustrated in Figure 4D, a combination of 13 µmol/L LA and 5 µmol/L VER potentiated ADR-induced cytotoxicity in Bcap37/MDR1 cells, with inhibitory rates up to 35%–50% compared to LA or VER alone treatment.
Discussion
The treatment of human cancer has been seriously hampered for decades by resistance to chemotherapeutic drugs[17]. Mechanisms underlying this resistance are far from being entirely known. The major hypothesis involves the existence of some cellular activities that tumor cells hijack to overcome the effects of cytotoxic drugs. Both biochemical and microenvironmental factors are involved in the mechanisms responsible for drug resistance. Between the cellular mechanisms, those mediated by proteins belonging to the family of ABC transporters are best known. These proteins are P-gp, the multi-drug resistance associated protein-1, and the breast cancer resistance protein. The ABC transporters are associated with drug efflux pumps and have been associated with MDR. Their treatment causes MDR by decreasing intracellular drug retention or by altering the target distribution of the drugs. New data suggest that the linkage of some of these proteins (e.g. P-gp) with the actin cytoskeleton may be of importance for their activity[18]. Thus, it seems that the function of these proteins may depend on a more complex cellular mechanism that involves more than a cellular structure. Although these proteins need to be characterized further, their significance as membrane efflux pump-related proteins in the resistance of human tumors to cytotoxic drugs has yet to be clarified.
In this study, the problem of MDR of human tumors has been faced by the use of an extract from Angelicae sinensis, an important herb in Chinese traditional medicine. Some formulas containing Angelicae sinensis, such as Siwu mixture, have been used in clinics as an auxiliary chemotherapy and have demonstrated reversing multidrug resistance in P-gp overexpressing human leukemia cells[19]. Based on the extraction of the mixture formula, it suggested the activation component might be a polysaccharide[20]. In our previous study, however, we found that the chloroform extracts of Angelicae sinensis had a more potent reversing effect than that of the polysaccharide fraction. Through step-by-step extracting and screening, we finally found that the dimmer of (Z)-ligustilide (LA) had the potential to reverse multidrug resistance in a variety of P-gp overexpressing MDR cells which were selected by increased doses of ADR (e.g. K562/ADR and MCF-7/ADR) or vincristine (e.g. KBv200). In order to further investigate the reversing effect of LA on P-gp-mediated drug resistance, we used Bcap37/MDR1 cells which were derived from the Bcap37 cells transfected with the human MDR1 gene[11]. With this model, we demonstrated that LA at subcytotoxic doses significantly reversed MDR by enhancing ADR- or VCR-induced G2/M arrest and apoptosis (Figures 2,3). Its reversing capacity was associated with its inhibitory effect on P-gp function (Figure 4). Both ADR and VCR are substrates of P-gp[6]. Typical P-gp modulators, such as VER and CsA, reverse MDR by competitively binding to P-gp. However, the effective doses of these drugs have been proven to induce severe side-effects in the treatment of cancer patients[6]. The results of our study have shown that LA behaves differently to the typical P-gp substrates. First, unlike VER, LA shows a similar IC50 between P-gp-negative and -overexpressing cells (Table 1), suggesting it is not a P-gp substrate. However, cotreatment with LA and VER synergistically enhances either ADR- or VCR-induced cytotoxicity (Figure 4).
In conclusion, our data demonstrate for the first time that LA, a component of Angelicae sinensis, can modulate P-gp function and overcome P-gp-mediated MDR in Bcap37/MDR1 cells, indicating that LA could be a highly feasible candidate for the development of a new P-gp modulator. Moreover, LA and VER cotreatment can synergistically inhibit P-gp function and enhance ADR-induced cytotoxicity, suggesting that a combination of LA and VER may be possible to develop a more sufficient with less toxic anti-MDR method.
References
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene 2003;22:7468-85.
- Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem 1989;58:137-71.
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48-58.
- Tarasova NI, Seth R, Tarasov SG, Kosakowska-Cholody T, Hrycyna CA, Gottesman MM, et al. Transmembrane inhibitors of P-glycoprotein, an ABC transporter. J Med Chem 2005;48:3768-75.
- Pec MK, Aguirre A, Fernandez JJ, Souto ML, Dorta JF, Villar J. Quantitative immunocytochemical assays of P-glycoprotein in breast carcinomas: correlation to messenger RNA expression and to immunohistochemical prognostic indicators. J Natl Cancer Inst 1994;86:1539-45.
- Kang Y, Perry RR. Effect of alpha-interferon on P-glycoprotein expression and function and on verapamil modulation of doxorubicin resistance. Cancer Res 1994;54:2952-8.
- Tang JC, Zhang JN, Wu YT, Li ZX. Effect of the water extract and ethanol extract from traditional Chinese medicines Angelicae sinensis (Oliv.) Diels, Ligusticum chuanxiong Hort. and Rheum palmatum L. on rat liver cytochrome P450 activity. Phytother Res 2006;20:1046-51.
- Lian N, Liu JB, Yan QM. Effect of jiawei donggui buxue decoction on the survival time of mice with cancer. Chin J Clin Rehabilitation 2004;8:5736-7.
- Lian N, Liu YM, Liu Y. Effect on modified Dang-gui Bu-xue decoction to curative effect of tumor treated by chemotherapy. J Chengdu Univ TCM 2003;26:18-9.
- Li BH, Lian N. Clinical observation on synergia and attenuation of Jia-wei Dang-gui bu-xue decoction to radiotherapy and chemotherapy of tumor: attachment report of 392 cases. J Chengdu Univ TCM 2005;28:7-9.
- Wang Y, Cao J, Zeng S. Establishment of a P-glycoprotein substrate screening model and its preliminary application. World J Gastroenterol 2004;10:1365-8.
- Su DM, Yu SS, Qin HL. New dimeric phthalide derivative from Angelica sinensis. Acta Pharm Sin 2005;40:141-4.
- Liu L, Ning ZQ, Shan S, Zhang K, Deng T, Lu XP, et al. Phthalide Lactones from Ligusticum chuanxiong inhibit lipopolysaccharide-induced TNF-alpha production and TNF-alpha-mediated NF-kappaB activation. Planta Med 2005;71:808-13.
- Deng S, Chen SN, Lu J, Wang ZJ, Nikolic D, van Breemen RB, et al. GABAergic phthalide dimers from Angelica sinensis (Oliv.) Diels. Phytochem Anal 2006;17:398-405.
- Augustin E, Mos-Rompa A, Skwarska A, Witkowski JM, Konopa J. Induction of G2/M phase arrest and apoptosis of human leukemia cells by potent antitumor triazoloacridinone C-1305. Biochem Pharmacol 2006;72:1668-79.
- Mullan PB, Quinn JE, Gilmore PM, McWilliams S, Andrews H, Gervin C, et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene 2001;20:6123-31.
- De Milito A, Fais S. Proton pump inhibitors may reduce tumour resistance. Expert Opin Pharmacother 2005;6:1049-54.
- Luciani F, Molinari A, Lozupone F, Calcabrini A, Lugini L, Stringaro A, et al. P-glycoprotein/actin association through erm family proteins: a role in p-glycoprotein function in human cells of lymphoid origin. Blood 2002;99:641-8.
- Xia L, Shen P. Studies on the effects of Siwu Mixture on reversing multidrug resistance of human erythrocyte leukemic cell line K562/ADM. Zhongguo Zhong Yao Za Zhi 2004;29:792-5.
- Shen P, Xia L, Zhang HJ. Discovery of the main active constituents of siwu mixture for reversing the multidrug resistance of human erythrocyte leukemic cell line K562/ADM. Zhongguo Zhong Yao Za Zhi 2005;30:520-3.