
Chronic palmitate exposure inhibits AMPKα and decreases glucose-stimulated insulin secretion from β-cells: modulation by fenofibrate1
Introduction
Adenosine monophosphate-activated protein kinase (AMPK), which serves as a metabolic master switch in response to alterations in cellular energy charge, has been reported to be implicated in the regulation of glucose and lipid homeostasis and insulin sensitivity[1–3]. It is activated by diverse stimuli that increase the AMP-to-ATP ratio (eg exercise and hypoxia) as well as by hormones (eg adiponectin and leptin)[4] and other antidiabetic agents (eg metformin and Thiazolidinediones)[5]. In peripheral tissues, AMPK activation by 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) or other activators of AMPK may be a selective tool to achieve normoglycemia by stimulating glucose uptake[6–8] in muscles and inhibiting hepatic glucose production[9,10]. In pancreatic β-cells, AMPK may modulate insulin secretion response to different concentration of glucose in INS-1 cells and isolated islets[11–13]. However, the role of AMPK in β-cells under chronic lipotoxic conditions is poorly understood.
Fenofibrate is an activator of the peroxisome proliferator-activated receptor-α (PPARα), which has been reported to upregulate genes of fatty acid β-oxidation pathways in various tissues[14,15]. Clinically, fenofibrate has been used for the treatment of dyslipidemia, mainly due to its ability to lower triglyceride levels, raise high-density lipoprotein levels, and decrease the levels of small, dense, low-density lipoprotein particles[16]. In otsuka long evans tokushima fatty (OLETF) rats, fenofibrate treatment may reduce adiposity, improve peripheral insulin action, and exert beneficial effects on pancreatic β-cells[17]. In primary human pancreatic islets, fenofibrate may prevent the fatty acid-induced impairment of glucose-stimulated insulin secretion (GSIS), apoptosis, and triglyceride accumulation[18]. In addition, PPARα activation was involved in insulin secretion in pancreatic β-cells[16,19–22], but whether and why the action is protective is still disputed.
In the present study, we aimed to investigate the effect of palmitate on AMPK expression and GSIS in isolated rat pancreatic islets and INS-1 β-cells, as well as the effect of fenofibrate on AMPK and GSIS in cells treated with palmitate.
Materials and methods
Rat pancreatic islet isolation and treatment Pancreatic islets were isolated from male Wistar rats weighing 230–275 g with collagenase solution followed by stationary in vitro digestion as previously reported[23]. The use of animals and the experimental protocols to which they were subjected were approved by the Institutional Animal Care and Use Committee of Shandong University (Jinan, China). Freshly isolated islets were then transferred to 24-well plates (10 islets per well) for the secretory experiment or a culture dish of 6 cm diameter (200 islets per dish) for Western blotting. They were cultured for 24 h in RPMI-1640 medium (Invitrogen, Grand island, NY, USA) containing 11.1 mmol/L glucose supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen, USA), 100 IU/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere (5% CO2 and 95% air) at 37 °C. Before commencing the experiment (24 h after seeding), the medium was replaced with freshly prepared RPMI-1640 containing 11.1 mmol/L glucose and supplemented with either bovine serum albumin (BSA; Sigma, St Louis, MO, USA) alone or BSA coupled to palmitate (0.2 or 0.4 mmol/L)[24],in the presence or absence of 5 µmol/L fenofibrate (a gift from Laboratories Fournier SA, Rue de Pres Potets, Fontaine-les-Dijon, France) for 48 h. The BSA-coupled palmitate was made in a molar ratio of 5:1.
Cell culture and treatment The rat insulinoma cell line INS-1 (at passages below 40) were grown in a monolayer as described previously[25] in the RPMI-1640 medium containing 11.1 mmol/L glucose supplemented with 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10% FBS, 1 mmol/L sodium pyruvate, 2 mmol/L L-glutamine, 50 µmol/L β-mercaptoethanol, 100 IU/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere (5% CO2 and 95% air) at 37 °C. The cells were seeded at 2×105 per well in 1 mL medium in a 24-well plate for secretory experiment, and at 1×106 per well in a 6-well plate for the RNA detection, and at 4×106 cells in a culture dish of 10 cm diameter for Western blotting. When the INS-1 cells were 80%–90% confluent, the medium was replaced with fresh medium and the treatment was the same as that of the isolated islets.
RNA isolation and cDNA synthesis The cultured cells or islets were harvested in TRIzol, and RNA was extracted according to a protocol of TRIzol isolation (Invitrogen, USA). The extracted RNA was resuspended in diethyl pyrocar-bonate-treated water and quantified by a DU640 nuclearic acid analyzer (Beckman, Fullerton, CA, USA). In total, 2 µg RNA was reverse-transcribed (RT) into the cDNA in a final reaction solution of 20 µL containing 5 mmol/L MgCl2, 2 µL 10×RT buffer, 1 mmol/L dNTP, 20 U RNase inhibitor, 5 U Avian myeloblastosis virus (AMV) reverse transcriptase, 2.5 µmol oligo(dT) primer, and 2 µg RNA and RNase-free dH2O using a RNA PCR kit (TaKaRa, Otsu, Shiga, Japan). The mixtures were heated as per the following conditions: 10 min at 30 °C, 30 min at 42 °C, 5 min at 99 °C, and 5 min at 5 °C. The extracted cDNA was used at real-time PCR or stored at -80 °C.
Real-time PCR Quantitative 3-step real time PCR was performed using Quantitect SYBR green kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions on an ABI 7700 prism real-time PCR instrument and software (Applied Biosystems, Branchburg, NJ, USA). The reaction volume was 25 µL and contained 12.5 µL 2×QuantiTect SYBR green PCR master mix, 0.5 µmol/L primers, and 100 ng cDNA and RNase-free water. The primers used for the PCR are detailed in Table 1. The reaction conditions were as follows: 1 cycle for 2 min at 50 °C, 1 cycle for 15 min at 95 °C, 40 cycles for 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. All quantifications were performed with rat GAPDH as an internal standard. The PCR products were visualized with gel electrophoresis to confirm a single product of the correct size (100 bp).
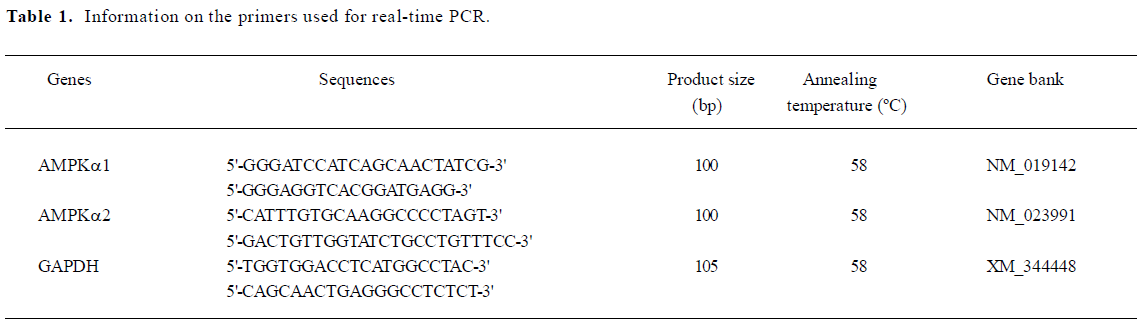
Full table
Protein analysis by Western blotting and enhanced chemiluminescence (ECL) detection The cultured islets and INS-1 cells were washed twice with ice-cold phosphate-buffered solution (PBS) and placed immediately in Radio immunoprecipitation assay (RIPA) lysis buffer containing 1×PBS, 1% nonylphenylpolyethylenglycol P-40 (NP-40), 0.1% SDS, 5 mmol/L EDTA, 0.5% sodium deoxycholate, 1 mmol/L sodium orthovanadate, and 1 mmol/L phenylmethyl sulfonylfluoride. The lysates were gently mixed for 10 min on ice and then centrifuged at 10 000×g for 10 min at 4 °C. The protein concentration of the extracts was determined according to the bicinchoninic acid (BCA) protein assay method using BSA as the standard. Then 60 µg of protein extracts were separated by 10% SDS-PAGE and electroblotted onto nitrocellulose membranes. The membranes were blocked with 5% non-fat milk in TBST (10 mmol/L Tris, 150 mmol/L NaCl, and 0.1% Tween 20) for 1 h and then incubated with the specific primary antibody of total AMPKα (T-AMPKα; at 1:1000 dilution, cell signaling, Danvers, MA, USA), phosphorylated AMPKα (P-AMPKα; cell signaling at 1:500 dilution), and phosphorylated acetyl coenzyme A carboxylase (P-ACC; cell signaling at 1:1000 dilution) overnight at 4 °C. After incubation with the relative second antibody, immune complexes were detected using the enhanced chemiluminescence (ECL) method (Amersham Biosciences, Little Chalfont, Buckinghamshire, United Kingdom); immunoreactive bands were quantified using Alphaimager 2200 (Alpha Innotech, San Leandro, CA, USA). Values were corrected with the absorbency of the internal control (β-actin).
Insulin secretion The cultured cells or islets were washed and pre-incubated for 30 min in Krebs-Ringer bicarbonate buffer (KRB) medium with the following composition: 143 mmol/L Na+, 5.8 mmol/L K+, 2.5 mmol/L Ca2+, 1.2 mmol/L Mg2+, 124.1 mmol/L Cl–, 1.2 mmol/L SO4 3-, and 25 mmol/L CO32- (pH 7.4), supplemented with 10 mmol/L HEPES, 0.2% BSA, and 3 mmol/L glucose. Upon completion of the incubation, the buffer was removed completely and replaced with fresh KRB containing 20 mmol/L glucose for an additional 20 min incubation. After 20 min incubation, the media were collected for measuring GSIS using an insulin radioimmunoassay (RIA) kit (Beijing Atom HighTech, Beijing, China). For the total protein extraction, the cells were homogenized in RIPA lysis buffer (Shenneng Bo Cai, Shanghai, China) containing 1×PBS, 1% NP-40, 0.1% SDS, 5 mmol/L EDTA, 0.5% sodium deoxycholate, and 1 mmol/L sodium orthovanadate. The protein concentration was determined by BCA assay (Bio-Rad, Hercules, CA, USA). The insulin level of the medium was normalized to its cellular protein content.
Data analysis All of the experiments were repeated at least 3 times. Values are given as mean±SD. Data were analyzed using one-way ANOVA. Significance was established at P<0.05.
Results
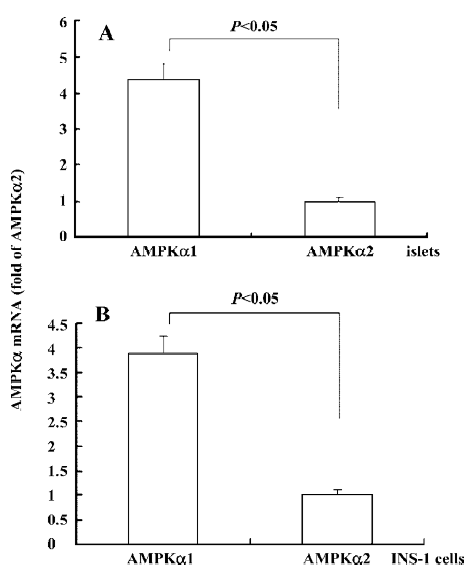
Expression of AMPKα1 and AMPKα2 in isolated rat islets and INS-1 cells The catalytic subunit of AMPK is the α subunit, which consists of 2 isoforms: α1 and α2. Real-time results showed that in the islets (Figure 1A) and INS-1 cells (Figure 1B), the mRNA expression of AMPKα1 was significantly higher than that of AMPKα2. The AMPKα1 expression was 4.37-fold in the islets and 3.89-fold in the INS-1 cells over that of AMPKα2, suggesting that AMPKα1 was the main isoform of AMPKα in the β-cells. Our result is consistent with a previous study of pancreatic β-cells[26].
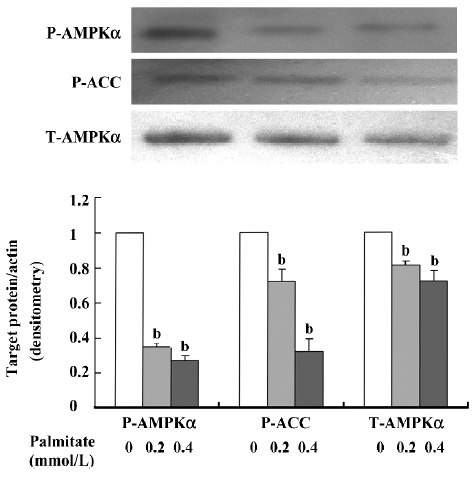
Inhibition effect of chronic exposure of β-cells to palmitate on AMPKα expression and activity To determine the effect of chronic exposure of β-cells to palmitate on AMPKα expression and activity, we cultured isolated rat pancreatic islets in RPMI-1640 medium containing 11.1 mmol/L glucose supplemented with and without 0.2 and 0.4 mmol/L palmitate for 48 h. The protein expression levels of T-AMPKα, P-AMPKα, and P-ACC were detected by Western blotting. The results demonstrated (Figure 2) that chronic exposure of isolated rat pancreatic islets to palmitate induced a significant decrease in P-AMPKα expression by 65% at 0.2 mmol/L palmitate and by 73% at 0.4 mmol/L palmitate treatment (P<0.05) in a dose-dependent manner. Accordingly, the expression of P-ACC, a downstream signal of AMPK, was also reduced (P<0.05), indicating that chronic palmitate exposure may inhibit the expression and activity of AMPKα.
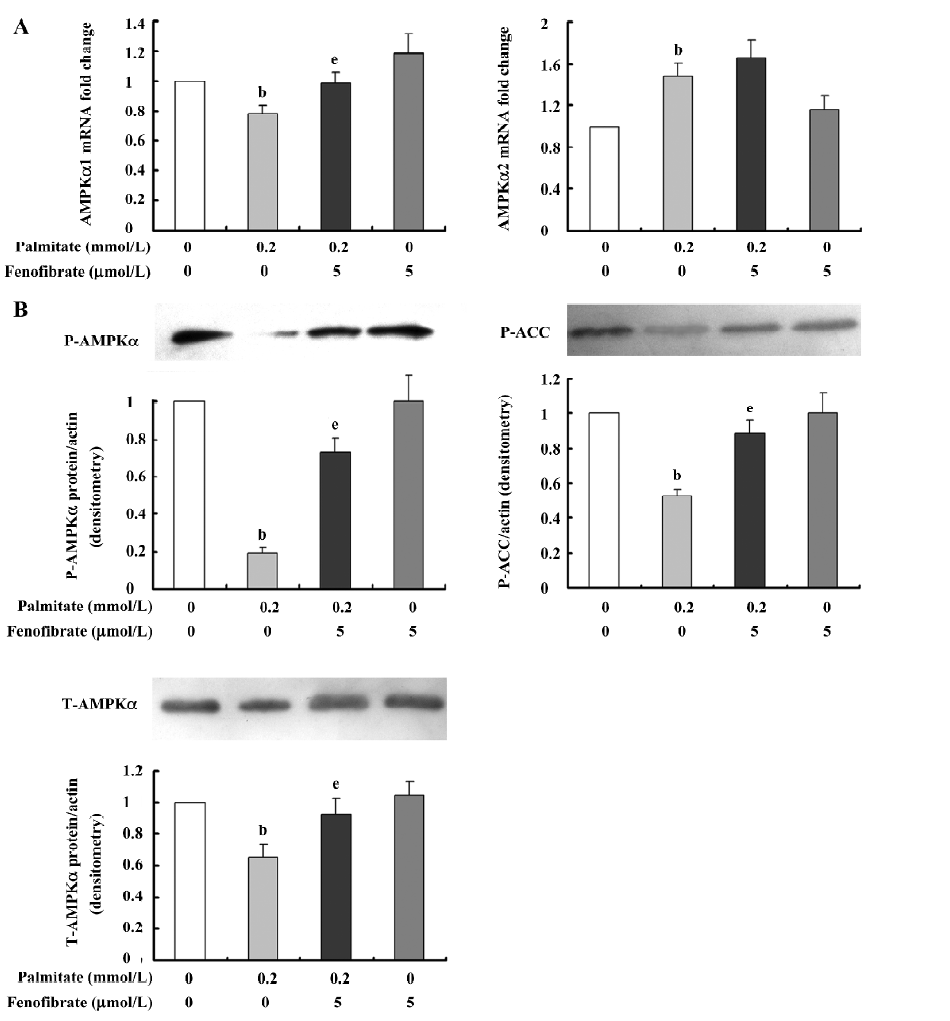
Amelioration effect of fenofibrate on AMPKα expression and activity in β-cells chronically exposed to palmitate To observe the effect of fenofibrate on AMPKα expression and activity in β-cells chronically exposed to palmitate, we cultured the INS-1 cells in RPMI-1640 medium supplemented with and without 0.2 mmol/L palmitate in the presence or absence of 5 µmol/L fenofibrate for 48 h. Then the mRNA levels of the AMPKα1 and α2 isoforms were measured by real-time PCR; the protein levels of T-AMPKα, P-AMPKα, and P-ACC and were detected by Western blotting. The results (Figure 3A) showed that in the palmitate-treated INS-1 cells, AMPKα1 mRNA expression decreased by 22% (P<0.05) while the AMPKα2 mRNA level was enhanced compared with the control. Compared with the palmitate-treated cells, the AMPKα1 mRNA level was enhanced by 26% (P<0.05) while there was no change to the cells treated with palmitate and fenofibrate together. Furthermore, the protein expression (Figure 3B) of T-AMPKα decreased by 47% in the palmitate-treated cells. Accordingly, the protein expressions of P-AMPKα and P-ACC were reduced respectively by 34% and 81% (P<0.05) in the palmitate-treated cells, respectively. Compared with the palmitate-treated cells, the cells treated with fenofibrate and palmitate together showed a remarkable increase in the protein expression of T-AMPKα by 40%, P-AMPKα by 68%, and P-ACC by 68% (P<0.05).
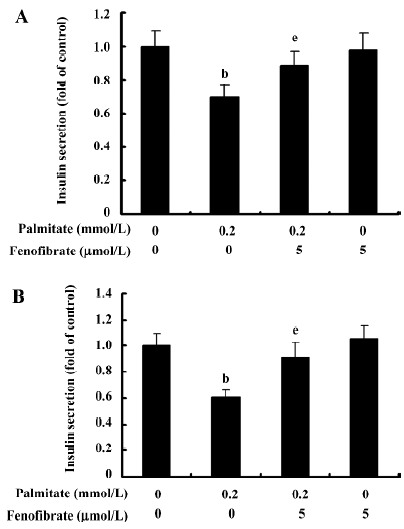
Effect of chronic exposure of α-cells to palmitate and fenofibrate on glucose-stimulated insulin secretion Insulin secretion was measured in the isolated islets and INS-1 cells by RIA induced by 20 mmol/L glucose as GSIS. As shown in Figure 4, GSIS was markedly reduced by 30% in the isolated islets pretreated for 48 h with 0.2 mmol/L palmitate. Accordingly, in the palmitate-treated INS-1 cells, GSIS decreased by 40%, implying the impairment role of palmitate on insulin secretion. Compared with the palmitate-treated islets and INS-1 cells, GSIS was restored to normal in the islets and INS-1 cells pretreated with palmitate and fenofibrate, indicating an improvement effect of fenofibrate on insulin secretion.
Discussion
In the present study, we demonstrated that chronic exposure of rat pancreatic β-cells to elevated palmitate reduced the expression of AMPKα and activity and impaired GSIS. Fenofibrate may potentiate GSIS associated with enhanced AMPKα expression.
AMPK is composed of 3 subunits: α, β, and γ. Among them, the α subunit is the catalytic subunit, which contains mainly 2 isoforms: α1 and α2. In our study, we first detected the mRNA levels of AMPKα1 and AMPKα2. The results revealed that not only AMPKα1 is more abundant than AMPKα2 in the isolated islets and INS-1 cells, but in the effect on the expression of AMPKα, the α1 isoform is predominant. Our result is consistent with the study of da Silva Xavier et al[26], in which an exclusive cytosolic localized for the α1 isoform and weak staining was present for α2 both in the cytosol and nucleus.
Next, we observed the effect of palmitate on AMPKα expression and activity as well as GSIS. Our study found that chronic exposure of islets and INS-1 cells to palmitate decreased the expression and activity of AMPKα and inhibited GSIS, indicating a possible role of AMPKα in β-cell lipotoxicity. In accordance with our findings, Liu et al[27] demonstrated that AMPKα expression and activity was decreased in the skeletal muscles of rats on a high-fat diet. The decrease of AMPK activity may activate ACC via dephos-phorylation, leading to an increase in the concentration of the product of ACC, malony-CoA, and the reduction in the carnitine palmitoyltransferase 1 expression to impair cellular fatty acid oxidation and accordingly attenuate insulin secretion. This suggests that the inhibition of AMPK expression and activity could play a role in insulin release. However, a recent study[28] found that chronic exposure of MIN6 cells to elevated palmitate for 24 h showed a sustained phosphorylation of AMPK. The results of the study appear to contradict those of ours, but can be explained by the difference in culture circumstances, such as different glucose concentrations and insufficient palmitate treatment time. Our results, at least partly, suggest that the decrease of AMPKα may be one of the mechanisms of β-cell lipotoxicity.
As already known, fenofibrate is a PPARα synthesis agonist. In several insulin-resistant rodent models, the administration of the PPARα agonist was reported to improve β-cell function[17]. Moreover, under conditions of lipotoxicity induced by chronic fatty acid exposure, different PPARα agonists significantly improved insulin secretion and the stimulation index in primary human islets[29,30]. In clinical experiments, fenofibrate can improve insulin secretion in hypertriglyceridemic individuals[31]. In addition, the activation of PPARα by the overexpression of PPARα/retinoid X receptor α potentiated GSIS in the rat islets and INS-1E rat β-cell line[19]. Our study supports the above view that PPARα activation can improve insulin secretion under conditions of lipotoxicity and demonstrates that fenofibrate restored GSIS impaired by palmitate in INS-1 cells, implying a beneficial role in β-cell function under pathological conditions of lipotoxicity.
However, we also observed that the improvement of insulin secretion is accompanied by AMPKα expression enhancement, implying a possible relationship between fenofibrate improving insulin secretion and AMPK activation. A similar relationship was found in human umbilical vein endothelial cells by Murakami et al[32], but they demonstrated that fenofibrate activating AMPK was unrelated to the effect of PPARα. Although in our study we did not verify whether the effect of fenofibrate on AMPKα was related to PPARα, it at least partly provides evidence that the promotion of fenofibrate on β-cell insulin secretion is associated with the expression of AMPKα. In addition, our study suggests a beneficial role of AMPKα activation in the insulin secretion of INS-1 cells under lipotoxic conditions. Some studies[27,33] have reported that AMPKα activation stimulated by metformin, AICAR, thiazolidinediones, leptin and so on may obviously ameliorate high-fat-induced insulin resistance and improve β-cell function. Diraison et al[34] proved that AICAR, an agonist of AMPK, can reverse in part the effects of sterol regulatory element binding protein-1c (SREBP1c) overexpression on triacylglycerol accumulation in transduction with SREBP1c of primary rat islets, suggesting that AICAR may act under conditions of β-cell lipid loading to favor the preservation of β-cell function. Although some studies suggest the activation of AMPK by AICAR (or by any other means) to exert deleterious or inhibitory effects on β-cells[26,35], the potential interpretation of these studies is the difference in the glucose concentrations and relative action time of AICAR.
In conclusion, our present study demonstrates that the α1 isoform expression, AMPKα expression and activity, and GSIS decreased in rat β-cells under palmitate-induced lipotoxic conditions. This disturbance could be ameliorated by fenofibrate, which is associated with the enhanced expression of AMPKα. Thus, our results suggest the effect of AMPKα on insulin secretion in β-cells treated with palmitate and the novel role of fenofibrate in improving insulin secretion is associated with AMPKα activation.
Acknowledgements
The authors thank Professor Xiao HAN for providing us with the INS-1 cells, and also the teachers at the Science Center of Shandong Provincial Hospital (Ji-nan, China) for excellent technical assistance.
References
- Bergeron R, Previs SF, Cline GW, Perret P, Russell RR, Young LH, et al. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 2001;50:1076-82.
- Fisher JS, Gao J, Han DH, Holloszy JO, Nolte LA. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 2002;282:E18-23.
- Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes 2002;51:2886-94.
- Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 2004;3:340-51.
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002; 277: 25 226–32.
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5' AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998;47:1369-73.
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 1997;273:E1107-12.
- Bergeron R, Russell RR, Young LH, Ren JM, Marcucci M, Lee A, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 1999;276:E938-44.
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167-74.
- Wollen N, Bailey CJ. Inhibition of hepatic gluconeogenesis by metformin. Synergism with insulin. Biochem Pharmacol 1988;37:4353-8.
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the α2 isoform. Biochem J 1998;334:177-87.
- Akkan AG, Malaisse WJ. Insulinotropic action of AICA riboside. I. Insulin release by isolated islets and the perfused pancreas. Diabetes Res 1994;25:13-23.
- Salt IP, Johnson G, Ashcroft SJ, Hardie DG. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic β cells, and may regulate insulin release. Biochem J 1998;335:533-9.
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J Biol Chem 1998;273:5678-84.
- Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, et al. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor α-deficient mice. J Clin Invest 1998;102:1083-91.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998;98:2088-93.
- Koh EH, Kim MS, Park JY, Kim HS, Youn JY, Park HS, et al. Peroxisome proliferator-activated receptor (PPAR)-α activation prevents diabetes in OLETF rats: comparison with PPAR-α activation. Diabetes 2003;52:2331-7.
- Lalloyer F, Vandewalle B, Percevault F, Torpier G, Kerr-Conte J, Oosterveer M, et al. Peroxisome proliferator-activated receptor-α improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes 2006;55:1605-13.
- Ravnskjaer K, Boergesen M, Rubi B, Larsen JK, Nielsen T, Fridriksson J, et al. Peroxisome proliferator-activated receptor α (PPARα) potentiates, whereas PPARα attenuates, glucose-stimulated insulin secretion in pancreatic β-cells. Endocrinology 2005;146:3266-76.
- Zhou YT, Shimabukuro M, Wang MY, Lee Y, Higa M, Milburn JL, et al. Role of peroxisome proliferator-activated receptor-α in disease of pancreatic β cells. Proc Natl Acad Sci USA 1998;95:8898-903.
- Tordjman K, Standley KN, Bernal-Mizrachi C, Leone TC, Coleman T, Kelly DP, et al. PPARα suppresses insulin secretion and induces UCP2 in insulinoma cells. J Lipid Res 2002;43:936-43.
- Holness MJ, Smith ND, Greenwood GK, Sugden MC. Acute (24 h) activation of peroxisome proliferator-activated receptor-α (PPARα) reverses high-fat feeding-induced insulin hypersecretion in vivo and in perfused pancreatic islets. J Endocrinol 2003;177:197-205.
- Gotoh M, Maki T, Satomi S, Porter J, Bonner-Weir S, O’Hara CJ, et al. Reproducible high yield of rat islets by stationary in vitro digestion following pancreatic ductal or portal venous collagenase injection. Transplantation 1987;43:725-30.
- Busch AK, Cordery D, Denyer GS, Biden TJ. Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic β-cell function. Diabetes 2002;51:977-87.
- Assimacopoulos-Jeannet F, Thumelin S, Roche E, Esser V, McGarry JD, Prentki M. Fatty acids rapidly induce the carnitine palmitoyltransferase I gene in the pancreatic β-cell line INS-1. J Biol Chem 1997;272:1659-64.
- da Silva Xavier G, Leclerc I, Varadi A, Tsuboi T, Moule SK, Rutter GA. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J 2003;371:761-74.
- Liu Y, Wan Q, Guan Q, Gao L, Zhao J. High-fat diet feeding impairs both the expression and activity of AMPKα in rats’ skeletal muscle. Biochem Biophys Res Commun 2006;339:701-7.
- Wang X, Zhou L, Li G, Luo T, Gu Y, Qian L, et al. Palmitate activates AMP-activated protein kinase and regulates insulin secretion from β cells. Biochem Biophys Res Commun 2007;352:463-8.
- Dubois M, Kerr-Conte J, Gmyr V, Bouckenooghe T, Muharram G, D’Herbomez M, et al. Non-esterified fatty acids are deleterious for human pancreatic islet function at physiological glucose concentration. Diabetologia 2004;47:463-9.
- Yaney GC, Corkey BE. Fatty acid metabolism and insulin secretion in pancreatic β cells. Diabetologia 2003;46:1297-312.
- Liang Z, Yan L, Li D, Li F, Qi Y, Cai M, et al. Impact of fenofibrate on insulin resistance and insulin secretion in individuals with hypertriglyceridemia. Chin J Endocrinol Metab 2007;23:8-11.
- Murakami H, Murakami R, Kambe F, Cao X, Takahashi R, Asai T, et al. Fenofibrate activates AMPK and increases eNOS phosphorylation in HUVEC. Biochem Biophys Res Commun 2006;341:973-8.
- Chen M. Lipotoxicity and AMPKα key target for its prevention and treatment. Chin J Endocrinol Metab 2007;23:5-7.
- Diraison F, Parton L, Ferre P, Foufelle F, Briscoe CP, Lexlerc I, et al. Over-expression of sterol-regulatory-element-binding protein-1c (SREBP1c) in rat pancreatic islets induces lipogenesis and decreases glucose-stimulated insulin release: modulation by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR). Biochem J 2004;378:769-78.
- Zhang S, Kim KH. Glucose activation of acetyl-CoA carboxylase in association with insulin secretion in a pancreatic β-cell line. J Endocrinol 1995;147:33-41.