
Combinatorial strategies for cancer eradication by silibinin and cytotoxic agents: efficacy and mechanisms1
Introduction
Mustard gas, the first used drug for cancer chemo-therapy, was used as a chemical warfare agent during World War I and was reported to cause severe toxic effects, one being the suppression of bone marrow leading to low white blood cell counts[1]. Studies thereafter with other forms of mustards confirmed the antineoplastic action of nitrogen mustard and thus launched the new era of cancer chemotherapy[1]. The majority of chemotherapeutic drugs can be broadly classified[2] into: alkylating agents (nitrogen mustards, ethyleneimines, methylmelamines, methylhydra-zine derivatives, alkyl sulfonates, nitrosoureas, triazenes, and platinum compounds); antimetabolites (folic acid, pyrimidine, and purine analogs); natural products (vinca alkaloids, taxanes, epipodophyllotoxins, camptothecins, anthracycline antibiotics, and anthracenedione); hormones and antagonists (adrenocortical suppressants, adrenocorticosteroids, progestins, estrogens, anti-estrogens, androgens, anti-androgens, gonadotropin-releasing hormone analogs, and aromatase inhibitors); biological response modifiers; differentiating agents; proteasome inhibitors; substituted urea; tyrosine kinase inhibitors; and monoclonal antibodies. This review intends to focus on the combination studies (both in vitro and in vivo) of silibinin, an established cancer chemo-preventive agent with the 2 classes of antineoplastic agents: the platinum compounds belonging to the class of alkylating agents and the anthracycline antibiotics.
Alkylating agents
The chemotherapeutic and cytotoxic effects of alkylating agents are directly related to the formation of carbonium ion intermediates or related transition complexes that alkylate DNA[3,4]. The potential of these drugs to induce cell death via interference with DNA integrity in fast proliferating tissues forms the basis for their therapeutic and toxic properties[3,4]. Platinum complexes do not alkylate DNA; however, they covalently bind to nucleophilic sites on DNA and share many pharmacological attributes to the alkylating agents and are thus included in this group[3,4]. In 1965, the platinum coordination complexes were first identified as potential antiproliferative agents based on a study which showed that a current delivered between platinum electrodes resulted in the inhibition of Escheria coli proliferation[3]. The inhibition of bacterial replication was later related to the formation of inorganic platinum-containing compounds in the presence of ammonium and chloride ions[3]. After this discovery, many platinum-containing compounds were synthesized and tested against different experimental tumor systems. Cisplatin and other members of the group, like carboplatin and oxaliplatin, have shown great clinical efficacy[5]. The platinum agents have broad antineoplastic activity against epithelial malignancies and have been used for the treatment of head and neck, bladder, esophagus, lung, colon, testicular, and ovarian cancers[4]. The platinum complexes inhibit telomerase (a specialized DNA polymerase expressed in malignant cells that mediates telomere extension at the 3' ends of telomere DNA) activity, react with DNA to form both intrastrand and interstrand cross-links, and the DNA adducts so formed inhibit DNA replication and transcription and lead to breaks and miscoding[4]. These adducts are recognized by p53 and other checkpoint proteins and result in the induction of apoptosis[5]. Malignant cells with mutant or absent p53 fail to arrest cell cycle progression and do not undergo apoptosis and are thus resistant to these drugs[5]. Other mechanisms that contribute to the resistance of the tumor to the platinum drugs are a reduction in drug accumulation in cancer cells due to barriers across the cell membrane, drug inactivation, alterations in drug target, and faster removal of the DNA adducts[4,5]. The cisplatin adducts are removed by nucleotide exchange repair, homologous recombinant repair, and translesion synthesis, which serve as cellular defense mechanisms against the toxic effects of the platinum drugs and thus contribute to tumor resistance to cisplatin[5].
Mismatch repair (MMR) increases the cytotoxicity of the cisplatin in tumor cells by the binding of the MMR complex to DNA adducts; dysfunction of this type of DNA repair also results in increased resistance of tumor cells to this drug[5]. The DNA adducts of cisplatin have been also shown to inhibit the anti-apoptotic function of NF-κB by inhibiting the binding affinity of NF-κB to the specific DNA elements[5]. Thus, NF-kB activation/inhibition is involved in the resistance/sensitivity of tumor cells to cisplatin. Carboplatin shows cross-resistance with cisplatin and forms similar types of DNA adducts, but with different sequence preferences[5].
The adverse reactions of cisplatin include renal toxicity, gastrointestinal toxicity, peripheral neuropathy, and ototoxicity[3]. Carboplatin is less toxic to kidneys and the nervous system; however, hematological adverse effects are more common[3].
Anthracyclines
The anthracycline antibiotics, another class of chemotherapeutic agents, are derived from the fungus Streptococcus peucetius[6,7]. They have a tetracyclic ring structure attached to the sugar, daunosamine[6,7]. Cytotoxic agents in this class have quinone and hydroquinone moieties on adjacent rings that permit the exchange of electrons[6,7]. These compounds intercalate with DNA and directly affect transcription and replication[6,7]. These drugs form a tripartite complex with DNA and topoisomerase II (an ATP-dependent enzyme that binds to DNA and produces double-strand breaks at the 3' phosphate backbone, allowing strand passage and the uncoiling of supercoiled DNA, after which it religates the DNA strands). The formation of the tripartite complex with anthracyclines inhibits the religation of the broken DNA strands and leads to apoptosis[2,7]. Thus, the defect in DNA double-strand break repair mechanisms results in cell damage causing cell cycle arrest and apoptotic death; however, an overexpression of transcription-linked DNA repair contributes to resistance to these drugs[7].
In addition to be topoisomerase II poison, the chemotherapy drugs in this class also possess potential for generating free radicals (by interaction with iron), thereby causing irreversible cardiomyopathy[8], which is related to the total dose of the drug. Doxorubicin also produces formaldehyde in iron-mediated free radical reactions; formaldehyde and doxorubicin then react to form a conjugate which intercalates with DNA by a process referred to as “virtual cross linking”[7]. Treatment with doxorubicin also results in malondialdehyde formation and DNA intercalation-oxopropenylation, which results in DNA damage[7]. Doxorubicin also activates p53–DNA binding which causes the induction of Cip1/p21 and results in the G1 arrest of p53-proficient cells[7]. The overexpression of p21 is also associated with resistance to doxorubicin as it is associated with increased G1 arrest that facilitates DNA repair before the cells undergo DNA replication[7]. The p53-deficient cells, not arrested at the G1 phase, progress through to the S phase where the expression of α-isoform of topoisomerase II is increased during DNA synthesis; these cells become sensitive to doxorubicin[7].
Another mechanism of resistance to doxorubicin involves the role of proteasome. Doxorubicin is transported to the nucleus via the complex formation with proteasome in the cytoplasm, thus in cells with increased nuclear sequestration of proteasome, there are reduced levels of transported doxorubicin and these cells show resistance to doxorubicin[7]. Doxorubicin causes the activation of NF-κB in cancer cells, which in turn inhibits apoptosis induced by doxorubicin; cells with increased activity of NF-κB are thus resistant to doxorubicin[7].
The clinical use of the drugs in this class is thus limited due to the development of resistance in tumor cells, toxicity to normal tissues, and adverse cardiac effects[7,8]. However, it is important to note that daunorubicin and idarubicin are effective in patients with acute leukemias, whereas doxorubicin and epirubicin have shown clinical efficacy against solid tumors in their clinical manifestations[7,8]. The structurally similar agent mitoxantrone (anthracenedione) is used in high-dose chemotherapy in advanced hormone-independent prostate cancer and acute myeloid leukemia. It causes less cardiac toxicity than the anthracyclines, and its toxicity includes myelosuppression and mucositis[9,10].
Anticancer effects via the modulation of cell cycle checkpoints
In order to understand how the anticancer agents eventually inhibit tumor cell growth or how a particular chemo combination would ultimately result in a synergistic effect, an understanding of cell cycle regulation is essential. The cell cycle is divided into 2 basic parts: mitosis and interphase. Mitosis (nuclear division) corresponds to the separation of daughter chromosomes and ends with cell division (cytokinesis). Interphase involves both cell growth and DNA synthesis in an orderly manner in preparation for cell division[11,12].
While the cell grows at a steady rate throughout inter-phase, DNA is synthesized only during a portion of interphase[11,12]. Cytokinesis is followed by the G1 phase that corresponds to the interval between mitosis and the initiation of DNA replication[11,12]. During the G1 phase, the cell is metabolically active and continuously grows, but does not synthesize DNA. G1 is followed by DNA synthesis (S) phase followed by an interval following the termination of DNA synthesis (G2) phase, during which cell growth continues and proteins are synthesized in preparation for mitosis and then the mitotic (M) phase[11,12]. Different cellular processes, such as cell growth, DNA replication, and mitosis, are coordinated during cell cycle progression by a series of checkpoints and feedback controls that regulate progression through various phases of the cell cycle and prevent entry into the next phase of the cell cycle until the events of the preceding phase have been completed[13].
There is a major cell cycle regulatory point that controls progression from G1 to S, and having passed this regulatory point, the cells are committed to entering the S phase and undergoing 1 cell division cycle even in the absence of further growth factor stimulation[14]. If appropriate growth factors are not available in G1, progression through the cell cycle stops at this point and the arrested cells then enter a quiescent stage of the cell cycle called G0, in which they are metabolically active but cease growth, have a reduced rate of protein synthesis, and can remain for a long period of time without proliferating until they receive external signals[13,14].
While the proliferation of most cells is regulated in G1, some cell cycles are controlled in G2, which prevents the initiation of mitosis until DNA replication is completed[14,15]. Progression is also arrested at the G2 checkpoint in response to DNA damage, which allows time for the damage to be repaired before being passed on to daughter cells[14,15]. DNA damage not only arrests the cell cycle in G2, but also arrests cell cycle progression in G1, which may allow repair of the damage to take place before the cell enters the S phase where the damaged DNA would be replicated[13–16]. Arrest at the G1 checkpoint is mediated by p53, which is rapidly induced in response to damaged DNA. It is important to emphasize here that mutations in the p53 gene are most common in human cancers[16]. Another important checkpoint occurs toward the end of mitosis, which monitors the alignment of chromosomes on the mitotic spindle and ensures that the chromosomes do not separate until a complete complement of chromosomes has been organized for the distribution of the daughter cells[12,15].
Cell cycle progression is regulated by the activity of cyclin-dependant kinases (Cdk) in association with their regulatory subunits known as cyclins as well as their negative regulators, which are named Cdk inhibitors[13]. The activation of different Cdk is specific to the distinct phases of the cell cycle; for example, the association of Cdk4/Cdk6 with D-type cyclins controls the early and mid-G1 phase progres-sion, whereas Cdk2–cyclin E regulates late G1 and G1–S transition, Cdk2–cyclin A for S-phase progression, and cell division cycle (Cdc)2 (also known as Cdk1)–cyclin B1 for G2–M phase transition (Figure 1)[13]. The activity of Cdk during cell cycle progression is regulated by different molecular mechanisms, where it first involves the association of Cdk with their cyclin partners, the formation of specific Cdk/cyclin complexes being controlled by cyclin synthesis and degradation[13]. To further elaborate on that as an example (Figure 1), cyclin B1 is a regulatory subunit required for the catalytic activity of the Cdc2 protein kinase. Cyclin B1 synthesis begins in the S phase, it then accumulates and forms a complex with Cdc2 throughout S and G2 during which Cdc2 is phosphorylated at 3 regulatory positions[13–16]. Phosphorylation at threonine-161 by Cdk-activating kinase (CAK) is required for Cdc2 kinase activity. The inhibitory phosphorylation of Cdc2 at tyrosine-15 and threonine-14 is catalyzed by protein kinase Wee1, which inhibits Cdc2 activity and leads to the accumulation of inactive Cdc2/cyclin B complexes throughout the S and G2 phases[13–16]. The dephosphorylation of Cdc2 at threonine-14 and tyrosine-15 by protein phosphatase Cdc25C causes the activation of the Cdc2/cyclin B complex and results in the transition from the G2 phase to the M phase[13–16]. Cell cycle arrest at this checkpoint is mediated by Chk2 and/or Chk1 kinase, activated by ataxia telangiectasia mutated (ATM) and ATM and Rad3 related (ATR), respectively, in response to DNA damage or incomplete replication[15]. Chk1 and Chk2 phosphorylate protein phosphatase Cdc25C, and thus prevent Cdc25C from dephosphorylating and activating Cdc2[15]. In the absence of such activation, however, the progression to mitosis is blocked and the cell remains arrested in G2[15]. Once activated, the Cdc2 protein phosphorylates a number of target proteins that initiate the events of the M phase[13–16]. The Cdc2 activity also triggers the degradation of cyclin B, which in turn inactivates Cdc2, leading the cell to exit from mitosis and undergo cytokinesis[13–16].
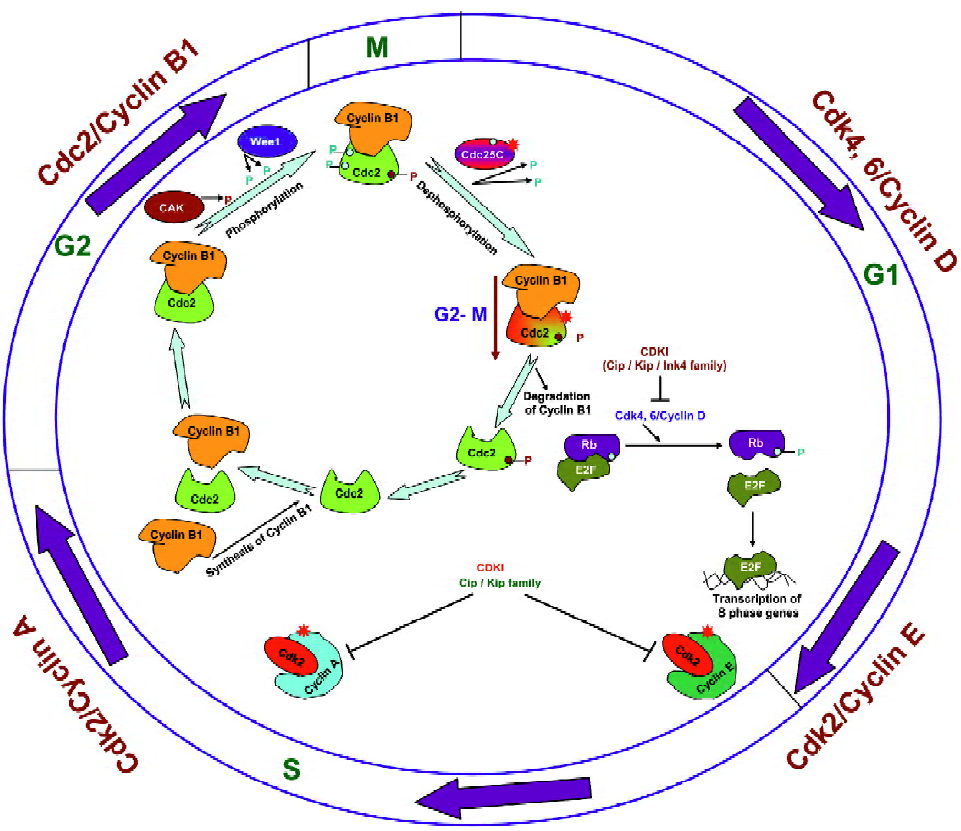
Apart from the regulation of their activity by phosphoryla-tion, the activity of Cdk could also be controlled by the binding of Cdk inhibitors to Cdk/cyclin complexes (Figure 1)[17]. Members of the Cip/Kip family regulate the progression through the G1 and S phases by inhibiting the activity of the complexes of Cdk2/4/6 with cyclins A, D, and E, and the members of the Ink4 family of Cdk inhibitors are specific for complexes of Cdk4 and 6 with cyclin D. The Ink4 inhibitors thus regulate progression only through the G1 phase[13,17]. Furthermore, retinoblastoma (Rb), a tumor suppressor gene whose inactivation leads to tumor development, is a key substrate protein of the Cdk4, 6/cyclin D complexes and is frequently mutated in a variety of human tumors[18]. Studies have revealed that the activity of Rb is regulated by changes in its phosphorylation status where it becomes hyperphos-phorylated by Cdk4, 6/cyclin D complexes as cells pass through the restriction point in G1 (Figure 1)[18]. In its hypo-phosphorylated form, Rb binds to the members of the E2F family of the transcription factors and thus acts as a repressor where the Rb/E2F complex suppresses the transcription of E2F-regulated genes to drive cell cycle progression to the next phase[18]. The phosphorylation of Rb by the Cdk4, 6/cyclin D complexes results in its dissociation from E2F; E2F then causes the activation of genes required for cell cycle progression[18]. E2F induces cyclin E and then Cdk2/cyclin E maintains Rb phosphorylation and this creates a positive feedback loop that is responsible for irreversible G1–S transition[17,18]. Cyclin A/B-dependent Cdk essential for S phase progression are subsequently activated and maintain the hyperphosphorylated form of Rb[17,18]. Upon withdrawal of mitogens, cyclin D synthesis stops and Cip/Kip proteins are available to bind to cyclin E/A–Cdk2, which blocks their catalytic activity and results in cell cycle arrest[17,18]. At this stage, in response to DNA damage, p53 stimulates the expression of the Cip1/p21 which inhibits several Cdk/cyclin complexes and thus inhibits Rb phosphorylation and directly inhibits DNA replication thereby stopping S phase progression[15,16,18].
Taking all these basic cell biology concepts together, it is reasonable to speculate that the efficacy of any anticancer agent would greatly depend upon its ability to target cell cycle checkpoints, either by the modulation of the cell cycle regulatory molecules (by direct or indirect action) thereby resulting in cell cycle arrest as a primary/ secondary effect or by targeting the damaged cells towards the end of their mitotic phase. The inhibition of cell growth is mediated either by arresting the cells in the G1 or S phase, bypassing the G2–M arrest via the G2–M checkpoint abrogation that would drive the cells to apoptosis, or by enhancing the G2–M checkpoint arrest[19]. The anticancer effects of drugs favor both the contrasting hypothesis: G2–M checkpoint abrogation or G2–M cell cycle arrest[20,21]. The mechanisms involved in both G2–M checkpoint abrogation or G2–M cell cycle arrest might be totally contrasting and conflicting, however, the endpoint of both these effects has been increased cytotoxicity[19].
Combined chemotherapy: a synergistic approach with “reduced toxicity”
Most chemotherapeutic drugs effectively target fast dividing cells causing their damage and are thus also referred to as “cytotoxic drugs”[22]. Consistent with this, the fast growing normal cells, such as those involved in hair growth and the mucosal lining of intestinal epithelium, are also effected leading to alopecia and intestinal complications. In addition, the chemotherapeutic drugs cause a wide variety of side–effects, like nausea, vomiting, anemia, malnutri-tion, immunosuppression, myelosuppression, hemorrhage, cardiotoxicity, hepatotoxicity, nephrotoxicity, ototoxicity, and non-specific neurocognitive problems[22].
The above listed toxic manifestations clearly suggest that the dosage of the drug is a critical factor; if the dose is too low, it will not be effective against the tumor, while at high doses the toxicity signs will be effective. Accordingly, the dosing schemes have been generated wherein the dose is adjusted according to the patient’s body surface area and blood volume[22]. Additionally, a number of strategies are being used in the administration of chemotherapeutic drugs, which include[22]: (1) combined modality chemotherapy that involves the use of drugs with other cancer treatments, like surgery and radiation therapy; (2) neoadjuvant chemotherapy or preoperative treatment where preceding surgery or radiation therapy, chemotherapeutic drugs are given to reduce the tumor size and make the local therapies more effective; (3) adjuvant chemotherapy or postoperative treatment, which involves continued drug intake even after surgery to reduce the risk of recurrence; (4) palliative chemotherapy that involves drug dosing only for decreasing the tumor load; it does not have any curative effects, but is expected to increase life expectancy slightly; and (5) combination chemotherapy, which involves treating a patient with a number of different drugs simultaneously.
In designing the specific regimens of combination chemotherapy for clinical use, a number of factors need to be considered; drugs should be most effective in combination, that is, their molecular/biochemical interactions should result in a synergistic response and thus it is more effective to combine drugs that do not share common mechanisms of activity and resistance and do not overlap in their major side-effects. In an effort to develop effective strategies that increase the therapeutic potential of anticancer drugs with less systemic toxicity, more strategies are being directed towards the investigation of dietary supplements and other phytotherapeutic agents (known for their high anticancer efficacy and low toxicity to normal tissues) for their synergistic efficacy in combination with anticancer drugs[23,24].
One such dietary agent is silibinin, which is a polyphenolic flavonoid isolated from the seeds of milk thistle (Silybum marianum). Both silibinin and its cruder form, known as silymarin, have clinically established hepatoprotective activity and have been used as dietary supplements against liver toxicity for over 3 decades[25]. Silibinin has shown promising chemopreventive and anticancer effects in various in vitro and in vivo studies[26–29]. Studies conducted by us and others have shown that silibinin and silymarin have anticancer activity against breast, skin, androgen-dependent and-independent prostate, cervical, bladder, hepatocellular, colon, ovarian, and lung cancer cells in culture and several in vivo animal cancer model systems[26]. The administration of these compounds to various laboratory animals has not shown any toxic or adverse effects in various acute, sub-chronic and chronic tests; there is no known LD50 for silymarin and silibinin in these animals[30,31]. This is also supported by our in vivo studies in animal models where we have used high doses of silibinin, up to 2 g/kg per d by oral gavage or 1%(w/w) in the diet, and observed no apparent signs of toxicity[27,29].
Combination studies of silibinin with chemo-therapeutic agents
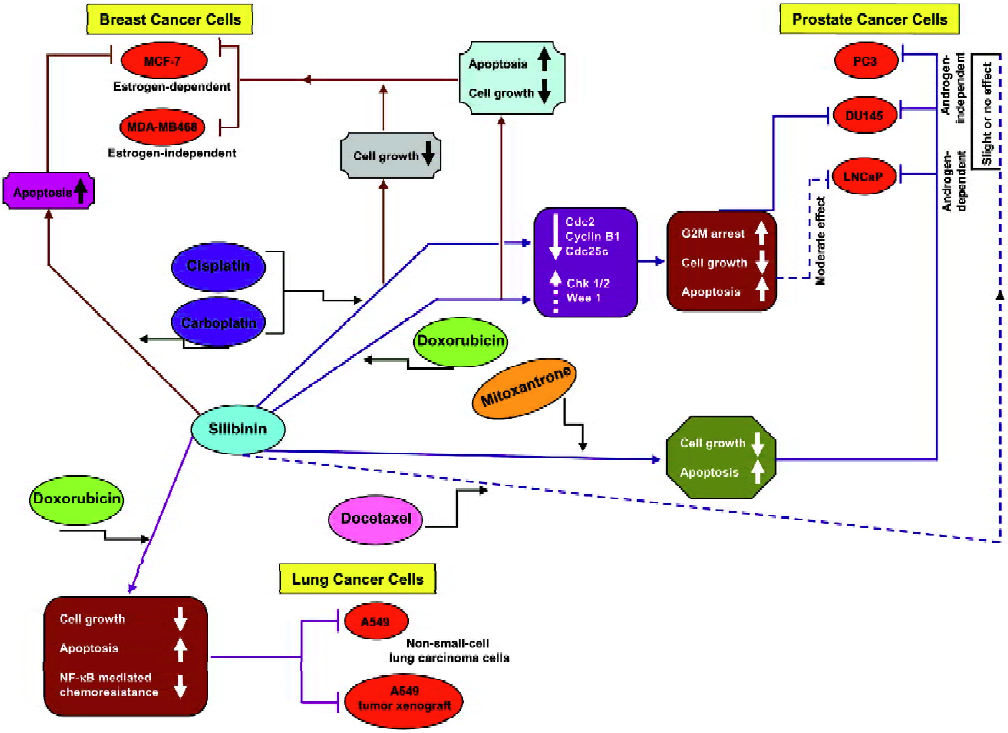
Several in vitro and in vivo combination studies of silibinin and chemotherapeutic drugs were carried out to analyze the effects of such a combination on growth inhibition, cell cycle regulation, and apoptosis. As summarized below, the 3 most important cancer systems, namely prostate, breast, and lung, have been investigated in detail-ed efficacy and mechanistic studies employing silibinin combination with chemotherapy agents (Figure 2).
Silibinin and chemo combination in prostate cancer
Studies by our group have shown that silibinin possesses strong anticancer efficacy against both androgen dependent and independent prostate carcinoma (PCa) cells in cell culture, where it inhibited cell growth and induced cell cycle arrest in LNCaP, PC-3, and DU145 cells[32–34]. Mechanistic studies conducted by us have also shown that the anticancer efficacy of silibinin against PCa cells is associated with the induction of differentiation morphology, a reduction in the prostate specific antigen level, the induction of cell cycle arrest accompanied by an increase in Cdk inhibitors, the inhibition of Cdk kinase activity, a decrease in the phosphorylation of Rb and Rb-related proteins, and their increased interaction with the E2F family of transcription factors[28]. We have also observed that silibinin inhibits the in vivo growth of DU145 xenografts in athymic nude mice mediated in part by an induction in the insulin-like growth factor-binding protein-3 level, the inhibition of proliferation and angiogenesis, and an induction in apoptosis[35]. Together these in vitro and in vivo studies clearly indicate that silibinin has strong efficacy against PCa, and therefore, our next goal was to assess whether synergistic action against PCa growth also exists when silibinin is used in combination with chemotherapeutic drugs.
Our chemo combination studies (Figure 2) showed that silibinin sensitizes the hormone refractory DU145 prostate carcinoma cells to cisplatin- and carboplatin-induced cell growth inhibition and apoptotic death[36]. The in vitro effects of silibinin and the platinum compounds, cisplatin (2 µg/mL) and carboplatin (20 µg/mL), on DU145 cells were assessed either alone or in combination. The findings of these studies revealed that the 48% cell growth inhibition observed with cisplatin increased to 50%–100% (P<0.05–0.001) when used in combination with 50–100 µmol/L silibinin. Similarly, the growth inhibition by carboplatin increased from 68% to 80%–90% (P<0.005–0.001) when used in combination with 50–100 µmol/L silibinin. The combination also resulted in a stronger G2–M arrest accompanied by a decrease in the expression levels of Cdc2, cyclin B1, and Cdc25C, which as earlier described, are essential for the transition from the G2 phase to the M phase of the cell cycle. Furthermore, compared to the effect of the single agent alone, there was a moderate increase in the Chk1 and Wee1 protein levels.
A similar chemo combination of silibinin and doxorubicin (Figure 2) also showed a strong synergism in the growth inhibitory effects in DU145 cells (combination index [CI]: 0.23–0.58). In this study, a dose-dependant effect of silibinin (10–100 µmol/L) and doxorubicin (10–100 nmol/L) on growth inhibition was determined followed by a combination of different doses of silibinin and doxorubicin, which resulted in a stronger synergistic growth inhibi-tion[37]. Among the combinations studied, a combination of 100 µmol/L silibinin and 25 nmol/L doxorubicin was most effective and showed an increase in growth inhibition of up to 80% compared to 43% and 53% when either of the agents were used alone. Further studies with this combination revealed that the synergistic effect was more profound when the cells were pretreated with either of the agents for 24 h followed by cotreatment with the other agent for another 24 h. Both cotreatments and simultaneous treatments showed an induction of G2–M arrest. Thus, regardless of the treatment regimen, the combination of silibinin and doxorubicin caused a strong G2–M arrest compared to G1 arrest by silibinin or a moderate G2–M arrest by doxorubicin alone. This G2–M arrest was also accompanied by a decrease in the expression levels of Cdc25C, Cdc2, and cyclin B1, together with a decrease in Cdc2 kinase activity and a moderate increase in Chk1/2 levels. There was also a ~3-fold increase in apoptosis when the 2 drugs were used in combination. Similar combination studies were also carried out in LNCaP cells, an androgen-dependant PCa cell line[37]. However, the synergistic effect was more profound in DU145 cells (the advanced prostate cancer cells) compared to the androgen-dependant LNCaP cells (Figure 2). Together, these studies showed that silibinin strongly synergizes the therapeutic effect of doxorubicin in advanced human prostate carcinoma DU145 cells via an induction of G2–M arrest and apoptotic cell death.
Combination studies of silibinin with docetaxel and mitoxantrone were also carried in PC-3, DU145, and LNCaP prostate cancer cell lines (Figure 2)[38]. A combination of silibinin (10–40 µmol/L) and docetaxel (2.5–5 nmol/L) exhibited little or no synergy in growth inhibitory and apoptotic effects in PC-3, DU145, and LNCaP cells, however, silibinin was able to overcome the relative resistance of PC-3 cells to mitoxantrone (25–200 nmol/L)-induced apoptosis. In this study, the combination of silibinin and mitoxantrone was also able to exhibit a synergistic action for apoptosis induction in DU145 and LNCaP cells and caused a synergistic decrease in the cell viability of all 3 cell lines.
Recently, we conducted a phase I clinical trial with silibinin in prostate cancer patients, in which 13 g of oral silybin–phytosome daily in 3 divided doses was administered to patients with advanced prostate cancer; each course was 4 weeks in duration[26]. An important observation in this study was that 100 µmol/L plasma level of free silibinin was achieved in the patients. This observation is highly significant with regards to the achievement of the physiological levels of a dose that was found to be highly effective in various in vitro studies including chemo combinations as discussed earlier.
Silibinin and chemo combination in breast cancer
We also assessed the synergistic effect of silibinin with chemotherapy agents in estrogen-dependant MCF-7 and -independent MDA–MB468 human breast carcinoma cell lines (Figure 2)[39]. In these cell lines, extensive dose- and time-dependant studies were initially carried out with the single agents followed by several combinations (10–100 µmol/L) of silibinin with doxorubicin (10–75 nmol/L), cisplatin (0.2–2 µg/mL), and carboplatin (2–20 µg/mL) to assess their synergistic/additive or antagonistic effect towards cell growth inhibition and apoptotic death. Combinations of these 3 drugs with silibinin showed strong synergistic effects on cell growth inhibition in both the cell lines, however, the strongest effect was due to a combination of 75 nmol/L doxorubicin and 100 µmol/L silibinin (CI: 0.35 for MCF-7 cells and CI: 0.45 for MDA–MB468 cells). A combination of silibinin and doxorubicin also showed strong apoptotic death in both the cell lines. The silibinin and carboplatin combinations increased the apoptotic effect in MCF-7 cells only, while the combination with cisplatin did not show any additional apoptotic effect in any of these cell lines.
Silibinin and chemo combination in lung cancer
Studies conducted by our group have also shown that silibinin strongly inhibits growth and induces apoptotic death in both human small cell lung carcinoma (SCLC) and non-small cell lung carcinoma (NSCLC) cells together with an alteration in cell cycle checkpoints[40]. Silibinin caused an increase in the G0–G1 population in SCLC SHP77 cells and an increase in the S phase in NSCLC A549 cells at lower treatment times followed by an increase in the G0–G1 phase at 72 h. Further studies in A549 cells showed that a combination of 25 nmol/L doxorubicin and 60 mmol/L silibinin increases cell growth inhibition to 85%, which was significantly higher than that caused by either agent alone (Figure 2)[41]. The combination also resulted in a ~37-fold increase in apoptosis, suggesting a synergistic effect on apoptotic cell death as well. In a follow up of these cell culture studies, we have also shown that oral feeding of silibinin (200 mg/kg body weight, 5 d/week for 33d) inhibits tumor growth, cell proliferation and angiogenesis and induces apoptosis in A549 tumor xenografts in nude mice. There was also a synergistic effect when silibinin was used in combination with doxorubicin (4 mg/kg body weight, intraperitoneally once a week on d 1, 8, 15, and 22; a total of 4 doses). Furthermore, there was also a reduction in doxorubicin-induced systemic toxicity in mice when the drugs were used in combination[41]. Other studies have also shown that there is an activation of NF-κB by doxorubicin, which was possibly responsible for the resistance of A549 cells to this drug[42,43]. Based on the results of our earlier studies where silibinin sensitized DU145 cells for TNFα-induced apoptosis by inhibiting NF-κB activation via an inhibition of IKKα kinase activity, which led to the increase in the cytoplasmic IκBα level and ultimately inhibited the nuclear translocation of the p65 and p50 subunits of NF-κB[44], we assumed that silibinin would also reverse the NF-κB-based chemoresistance of A549 cells to doxorubicin. We confirmed this assumption when the combination treatment of cells with doxorubicin and silibinin was able to decrease the activation of NF-κB by doxorubicin together with the retention of p65 and p50 in the cytosol by silibinin, which in turn significantly reduced the doxorubicin-induced resistance in A549 cells (Figure 2)[41]. The chemo combination with silibinin thus enhanced the therapeutic efficacy of doxorubicin towards the inhibition of lung tumor growth.
Conclusion
In the unfortunate scenario where the most effective cytotoxic drugs face limited clinical applications due to their high toxicity and increased chemoresistance, combination therapy/prevention is gaining increased attention as an effective alternative to increase therapeutic efficacy and minimizes the systemic toxicity of these chemotherapeutic agents. In this regard, the present review summarizes the effects of the combination of silibinin and chemotherapeutic drugs on growth inhibition, cell cycle regulation, and apoptosis induction in prostate, breast, and lung cancer cells. Together, the results indicate a synergistic effect of silibinin on growth inhibition, the reversal of chemoresistance, apoptosis induction, and a strong increase in G2–M checkpoint arrest when given in combination with these drugs. These results are highly significant with respect to the combined chemotherapy approach where the criteria for combination are that the response has to be synergistic and that the drugs should not share common mechanisms of resistance and not overlap in their major side-effects. Accordingly, we emphasize that silibinin is an ideal candidate to be used in clinical combination with the chemotherapeutic drugs which would increase their therapeutic efficacy and at the same time reduce the adverse effects associated with their high doses. How-ever, more extensive studies in this direction are needed in the near future, not only with silibinin, but also several additional non-toxic and naturally-occurring cancer chemopreventive agents to justify their potential clinical application in combination chemotherapy.
Footnote
Original studies in our silymarin/silibinin and cancer chemoprevention program are funded by NCI RO1 grants (N
References
- Hajdu SI. 2000 years of chemotherapy of tumors. Cancer 2005;103:1097-102.
- Goodman LS, Gilman A, Brunton LL, Lazo JS, Parker KL. Goodman & Gilman’s the pharmacological basis of therapeutics. 11th ed. New York: McGraw-Hill; 2006.
- Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol 2001;67:93-130.
- Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. Oncol Rep 2003;10:1663-82.
- Brabec V, Kasparkova J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist Updat 2005;8:131-46.
- Binaschi M, Bigioni M, Cipollone A, Rossi C, Goso C, Maggi CA, et al. Anthracyclines: selected new developments. Curr Med Chem Anticancer Agents 2001;1:113-30.
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracy-clines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004;56:185-229.
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyo-cyte apoptosis. Cancer Res 2000;60:1789-92.
- Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513-20.
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-12.
- Sherr CJ. Cell cycle control and cancer. Harvey Lect 2000;96:73-92.
- Cooper GM, Hausman RE. The cell: a molecular approach. 3rd ed. Washington, D C: ASM Press, Sinauer Associates; 2004.
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003;36:131-49.
- Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell 1997;88:315-21.
- Russell P. Checkpoints on the road to mitosis. Trends Biochem Sci 1998;23:399-402.
- Vousden KH. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta 2002;1602:47-59.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501-12.
- Genovese C, Trani D, Caputi M, Claudio PP. Cell cycle control and beyond: emerging roles for the retinoblastoma gene family. Oncogene 2006;25:5201-9.
- DiPaola RS. To arrest or not to G(2)-M Cell-cycle arrest: commentary re: A K. Tyagi et al. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin cancer res, 8: 3512-3519, 2002. Clin Cancer Res 2002;8:3311-4.
- Hirose Y, Berger MS, Pieper RO. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res 2001;61:5843-9.
- Shapiro GI, Supko JG, Patterson A, Lynch C, Lucca J, Zacarola PF, et al. A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res 2001;7:1590-9.
- Devita VT, Hellman S, Rosenberg RA. Cancer: Principles and Practice of Oncology. 7th ed. Pliladelphia-PA: Lippincott Williams and Wilkins; 2005.
- Kelloff GJ. Perspectives on cancer chemoprevention research and drug development. Adv Cancer Res 2000;78:199-334.
- Sporn MB, Suh N. Chemoprevention of cancer. Carcinogenesis 2000;21:525-30.
- Wellington K, Jarvis B. Silymarin: a review of its clinical properties in the management of hepatic disorders. BioDrugs 2001;15:465-89.
- Kaur M, Agarwal R. Silymarin and epithelial cancer chemopreven-tion: How close we are to bedside? Toxicol Appl Pharmacol (Published online Nov, 15th 2006).
- Singh RP, Agarwal R. Prostate cancer prevention by silibinin. Curr Cancer Drug Targets 2004;4:1-11.
- Singh RP, Agarwal R. Mechanisms of action of novel agents for prostate cancer chemoprevention. Endocr Relat Cancer 2006;13:751-78.
- Singh RP, Agarwal R. Prostate cancer chemoprevention by silibinin: bench to bedside. Mol Carcinog 2006;45:436-42.
- Vogel G, Trost W, Braatz R, Odenthal KP, Brusewitz G, Antweiler H, et al. Pharmacodynamics, site and mechanism of action of silymarin, the antihepatoxic principle from Silybum mar. (L) Gaertn. 1. Acute toxicology or tolerance, general and specific (liver-) pharmacology. Arzneimittelforschung 1975;25:82-9.
- Mereish KA, Bunner DL, Ragland DR, Creasia DA. Protection against microcystin-LR-induced hepatotoxicity by Silymarin: biochemistry, histopathology, and lethality. Pharm Res 1991;8:273-7.
- Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci USA 1999;96:7490-5.
- Zi X, Zhang J, Agarwal R, Pollak M. Silibinin up-regulates insulin-like growth factor-binding protein 3 expression and inhibits proliferation of androgen-independent prostate cancer cells. Cancer Res 2000;60:5617-20.
- Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene 2006;25:1053-69.
- Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res 2002;62:3063-9.
- Dhanalakshmi S, Agarwal P, Glode LM, Agarwal R. Silibinin sensitizes human prostate carcinoma DU145 cells to cisplatin- and carboplatin-induced growth inhibition and apoptotic death. Int J Cancer 2003;106:699-705.
- Tyagi AK, Singh RP, Agarwal C, Chan DC, Agarwal R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G2-M arrest, and apoptosis. Clin Cancer Res 2002;8:3512-9.
- Flaig TW, Su LJ, Harrison G, Agarwal R, Glode LM. Silibinin synergizes with mitoxantrone to inhibit cell growth and induce apoptosis in human prostate cancer cells. Int J Cancer 2007;120:2028-33.
- Tyagi AK, Agarwal C, Chan DC, Agarwal R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol Rep 2004;11:493-9.
- Sharma G, Singh RP, Chan DC, Agarwal R. Silibinin induces growth inhibition and apoptotic cell death in human lung carcinoma cells. Anticancer Res 2003;23:2649-55.
- Singh RP, Mallikarjuna GU, Sharma G, Dhanalakshmi S, Tyagi AK, Chan DC, et al. Oral silibinin inhibits lung tumor growth in athymic nude mice and forms a novel chemocombination with doxorubicin targeting nuclear factor kappaB-mediated inducible chemoresistance. Clin Cancer Res 2004;10:8641-7.
- Das KC, White CW. Activation of NF-kappaB by antineoplastic agents. Role of protein kinase C. J Biol Chem 1997;272:14914-20.
- Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med 1999;5:412-7.
- Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits constitutive and TNFalpha-induced activation of NF-kappaB and sensitizes human prostate carcinoma DU145 cells to TNFalpha-induced apoptosis. Oncogene 2002;21:1759-67.