
Biomarker and animal models for assessment of retinoid efficacy in cancer chemoprevention
Introduction
Vitamin A (retinol) is an essential dietary component. It is required for normal embryonic development, maintenance of growth and differentiation of epithelial cells, male reproductive activity, immune functions and night vision[1–5]. Classic studies by Howe and others[6–8] have established a connection between vitamin A and cell proliferation. Comprehensive studies covering the areas of epidemiology, molecular biology, animal models and clinical science have provided strong evidence that vitamin A, through its metabolites, has tumor suppressor functions. Interest in using vitamin A and its metabolites for the prevention or treatment of cancer began in the 1980s. There have been both successes and failures in this area.
In this review, I will first discuss vitamin A metabolism and molecular function. This will be followed by a summary of our current knowledge of altered vitamin A metabolism and molecular function in cancer cells. Next I will outline the progress in the use of biomarkers to assess the efficacy of retinoids in chemoprevention and cancer, from about 1994 to the present. This will be followed by discussion of various animal models that have or could be used to validate biomarkers. An elaboration of why animal studies are essential before clinical trials will be presented. Finally, I will draw some conclusions from published studies as to our current state of knowledge and suggest areas where high-priority targeted research is needed.
Vitamin A: metabolism and molecular action
We obtain vitamin A (retinol) from our diet because we lack the ability to synthesize this vitamin. Both beta carotene and retinyl esters serve as sources of retinol. Once absorbed by the intestine, retinol is esterified and transported by chylomicrons to the liver. Here it is stored by the stellate cells until it is needed to replenish vitamin A levels. Retinol leaves the liver conjugated to serum retinol binding protein, which, in turn, is bound to the plasma protein transthyretin. The pathway and components of retinol/retinoic acid action are illustrated in Figure 1. It was recently discovered that cells possess a plasma membrane protein called STRA6 that acts as a cell surface receptor for serum retinol binding protein[9]. This receptor also extracts retinol and delivers it to the inside of the cell for subsequent metabolism. Mutations in STRA6 produce a pleiotropic syndrome that resembles vitamin A deficiency[10]. This finding supports the essential role of STRA6 in retinoid biology. Once retinol is inside the cells, it is metabolized to retinaldehyde and then to retinoic acid. The first step is catalyzed by medium-chain alcohol dehydrogenases, short-chain retinol dehydrogenases and some members of the P450 family. The second step is catalyzed by retinol dehydrogenases and some members of the cytochrome P450 family.
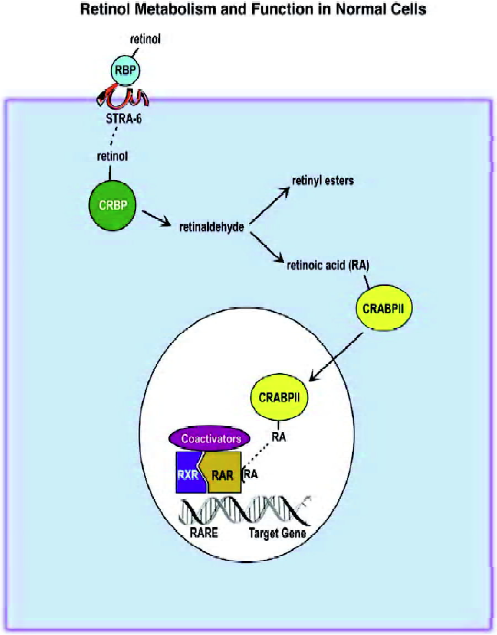
Two cytoplasmic retinol binding proteins (CRBP-I and -II) have been described. Likewise, there are two cytoplasmic binding proteins for retinoic acid (CRABP-I and -II). CRBP-II expression is limited to the adult small intestine, while CRBP-I is ubiquitously expressed. Its expression is stimulated by retinoic acid[11,12]. The expression of CRABP-II is somewhat limited, whereas CRABP-I is ubiquitously expressed[13]. Retinoic acid treatment induces CRABP-II expression[14]. The CRBP proteins appear to sequester retinol and shuttle it to different compartments, where it can be metabolized or stored as retinyl esters[15]. In contrast, type II CRABP delivers retinoic acid to the nucleus and can interact with nuclear retinoic acid receptors to alter their activity[16,17].
The most biologically active form of vitamin A is retinoic acid. It appears that most of the biological actions of retinoic acid are mediated by its nuclear receptors. These receptors have been found to contain the same structural modules as a family of steroid hormone receptors. Extensive screening of cDNA libraries revealed that there is a family of retinoic acid nuclear receptors. One class of receptors (RARα, β and γ) binds all-trans and 9-cis retinoic acid[18], whereas a related class of receptors (RXR α, β, and γ) only binds 9-cis retinoic acid with high affinity[19]. In addition, it is known that the RXR form heterodimers with a number of other nuclear receptors, such as RAR[20,21], vitamin D3 receptor[22,23], thyroid hormone receptor[24,25], peroxisomal proliferator-activator receptor[26,27] and a number of orphan receptors[28,29]. Under physiological conditions only the RXR:RAR heterodimer leads to productive DNA binding[30].
These ligand-activated receptors stimulate the expression of target genes through binding to specific retinoic acid response elements (RARE) usually located in the promoter region[31]. The consensus is that RARE is composed of a direct repeat of 5’PuG(G/T)TCA3’ separated by five other nucleotides[32]. However, there are a considerable number of RAR target genes whose RARE composition and location vary considerably from the consensus[33]. There are also some genes whose expression is apparently directly inhibited by retinoic acid. The mechanism for this retinoid action is not fully understood[34].
Altered vitamin A metabolism and function in cancer
A number of animal studies have demonstrated that vitamin A deficiency induces an increase in the number of spontaneous and chemically induced tumors[35–37]. The addition of “pharmacological” amounts of vitamin A to the diet reduced the incidence of chemically induced tumors in animals[38–40]. Human epidemiological studies found an inverse relationship between vitamin A/beta-carotene intake or plasma levels and the incidence of several types of cancers, such as lung[41], head and neck[42] and breast[43]. Treatment of acute promyelocytic leukemia patients with retinoic acid induces remission with high frequency. These cumulative studies provide strong evidence that vitamin A and its biologically active metabolites inhibit tumorigenesis. Therefore, in order for cancers to form, the cells must find a mechanism to subvert the normal biological activity of retinoids. Evidence for these changes is discussed below.
Vitamin A uptake and metabolism are altered in a number of different types of cancer cells (see Table 1). It has been noted that tumor cells have low levels of retinyl esters when compared with their normal counterparts. The enzyme responsible for this esterification, lecithin:retinol acyltrans-ferase (LRAT) is defective in some cancers, whereas in others, CRBP-I expression is lost[44–46]. In addition, retinoic acid is metabolized more rapidly in some tumor cells and the use of chemical inhibitors of the 4-hyroxylase enzyme increases retinoic acid concentration in tumor cells[47]. A number of studies have documented that RAR are altered in cancer cells. Acute promyelocytic leukemia is caused by a translocation of RAR-α resulting in a fusion protein, usually with the promyelocytic leukemia gene[48]. A great deal of experimental evidence implicates RAR-β as a tumor suppressor. Its expression has frequently been found to be reduced or silenced in numerous tumors[49–52]. Cancer cells, which are sensitive to retinoic acid treatment, demonstrate an upregulation of RAR-β when the cells are treated with this retinoid; resistant cancer cells fail to increase RAR-b expression[53]. There is little evidence that the RAR-β gene is lost or mutated in cancer cells. Instead, epigenetic mechanisms play a predominant role in inactivating the function or expression of RAR-β. Two orphan receptors, COUP-TF and nurr77, affect the expression of RAR-β. COUP-TF increases and nurr77 inhibits the expression of RAR-β[54,55]. The relative amounts of these two orphan receptors in cancer cells tracks with their effect on RAR-β expression, that is, low COUP-TF and high nurr77. The major mechanism for RAR-β silencing in cancer is DNA methylation, especially in the promoter region of this gene[56–58]. Treatment of these retinoic acid resistant cells with demethylating agents, such as 5-aza-2’-deoxycytidine, results in re-expression of RAR-β and acquisition of sensitivity to retinoic acid[59,60].

Full table
Retinoids and chemoprevention: biomarkers
Oral premalignancy has been the subject of a number of retinoid prevention trials. The lesions, leukoplakia, are easily monitored and sampled. In 1986, a short-term, high-dose isotretinoin trial was reported. For 3 months, 44 patients received either placebo or 1–2 mg/kg per d isotretinoin. After the cessation of treatment, the patients were monitored for an additional 6 months. Patients receiving isotretinoin had a 67% clinical response rate compared with 10% in patients on the placebo. However, the high amounts of isotretinoin resulted in toxicity and more than 50% of the initial responders relapsed within 2–3 months after the cessation of treatment[61]. Further variations on this protocol included high-dose isotretinoin induction followed by low-dose isotretinoin maintenance[62]. Progression during low-dose maintenance was only 8%. Three other randomized trials with retinoids and oral premalignancy also reported positive results[63–65]. These studies used clinical and histological measures of response. Lotan et al[66] used in situ hybridization to detect mRNA of RAR in biopsies of premalignant oral lesions. The amount of RAR-β RNA was low in the premalignant lesions and increased in the lesions that responded to isotretinoin treatment. Therefore, in oral premalignancy and other similar states in other tissues, RAR-β is likely to be an important biomarker for retinoid chemo-prevention studies.
Lung cancer has been another target of retinoid chemoprevention. A controlled trial of heavy smokers used the metaplasia index from bronchoscopy as an end-point. In a 6-month trial with isotretinoin using 87 subjects, there was a significant reduction in the metaplastic index in both isotretinoin and the placebo subjects[67]. Because of the variability of the metaplastic index, intermediate biomarkers are being sought to incorporate into these clinical trials.
Premalignant skin lesions have also been the target of retinoid chemoprevention studies. These lesions are readily observed and biopsied, making the assessment of chemopre-ventive activity easy to quantify. Several trials have found that topical[68,69] and systemic[70,71] retinoid treatment resulted in a decrease in the number of actinic keratoses. However, there were relatively small numbers of patients in these studies, and the effects were reversible. Further studies, involving xeroderma pigmentosum patients and renal transplant patients, who are at high risk of developing basal cell and squamous cell skin cancer, respectively, had significantly fewer skin cancers[72,73]. However, the study population was small and the effects were reversible after cessation of retinoid treatment. Several large-scale skin cancer chemoprevention studies using subjects at lower risk for skin cancer had varying results. One study found a lower incidence of primary squamous cell, but not basal cell, cancer[74], but the other three studies found no significant differences[75–77].
Cervical dysplasia can be followed quite easily and has been the subject of a variety of chemoprevention studies. Advanced trials from the University of Arizona found that tretinoin delivered via a collagen matrix for 1 year resulted in 50% of patients having a decrease in dysplastic lesions[78]. In a follow-up study, tretinoin was found to be more active in reversing moderate dysplasia, but had little effect in patients with severe dysplasia[79].
In these early studies (1980–1994), few attempts were made to identify intermediate biomarkers for retinoid chemoprevention activity. This was probably because of a lack of sensitive techniques/reagents that are needed to routinely quantify markers in relatively small amounts of clinical material. Attempts have been made to define putative bio-markers by examining genes/proteins that interact with or are part of the retinoid pathway in premalignant tissues. For example, Lawrence et al[80] found that RXR-α was overex-pressed in 66% and 88% of non-comedo DCIS and comedo DCIS lesions, respectively, which are associated with a >8-fold and >12-fold risk, respectively, of developing breast cancer. In contrast, only 8% of lesions that have only a small risk of developing breast cancer had overexpression of RXR-α. Whether the preneoplastic lesions that express high amounts of RXR-α will be more or less susceptible to retinoid treatment is yet to be investigated. Recently, human radial growth phase melanoma cells have been used as a surrogate for screening agents for melanoma prevention. These melanoma cells are the closest to normal melanocytes outside of dysplastic nevi. The markers used for these studies were N-cadherin and P-cadherin. Both 4-HPR and 9-cis retinoic acid had some effects on reversing the changes in marker expression after ultraviolet (UV)-B radiation of the cells[81]. It is not clear whether the chosen markers are specific for retinoid chemoprevention activity in melanoma.
A significant amount of progress has been made in defining biomarkers in upper aerodigestive tract cancer. Initiated cells have 9p and 3p loss, whereas mild dysplastic cells have 17p loss, telomerase activation and global DNA methylation. RAR-β loss and EGFR are hallmarks of moderate dysplasia, and severe dysplastic lesions have p53 mutation and increased expression of cyclin D1. Frank carcinoma lesions are associated with increased angiogenesis[82]. In retinoid chemoprevention trials, resistance was associated with abnormal p53 expression, increased degree of genomic instability and lack of RAR-β induction after treatment with 13-cis-retinoic acid[83]. However, none of these biomarkers have been validated and it is likely that multiple markers will need to be used to avoid the risk of basing the effectiveness of the chemoprevention agent on the wrong marker. Figure 2 illustrates possible biomarkers for assessing retinoid efficacy in premalignant cells.
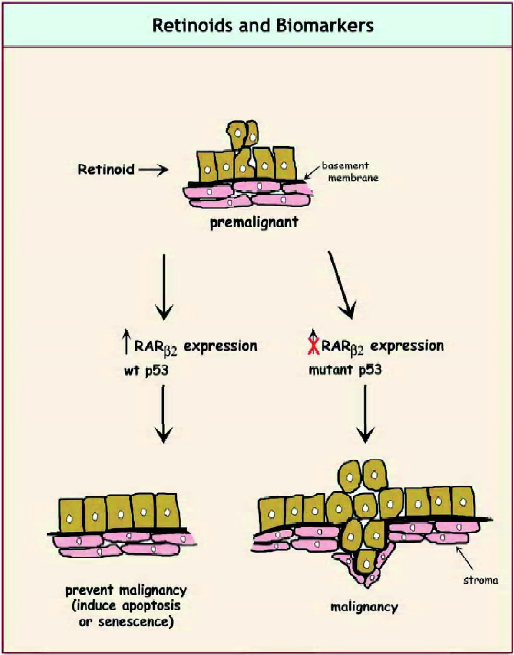
Global DNA methylation has been proposed as a bio-marker for chemoprevention in colon cancer[84]. Chemopre-vention agents that are effective in inhibiting the development of colon cancer decrease colonic DNA methylation, whereas those that are ineffective, including 9-cis retinoic acid, do not decrease colonic DNA methylation. Although there have been disappointing clinical primary prevention trials for beta carotene, alpha-tocopherol and retinyl palmitate, a limited trial of 13-cis retinoic acid, showed induction of RAR-β expression in lung cancer premalignancy[85]. Whether this will translate to effective chemoprevention of lung cancer in the subset of patients who express this marker is yet to be determined. The most promising marker for retinoid chemoprevention activity is induction of RAR-β expression. This receptor has been found to have reduced or no expression in cancers cells from breast, lung, head and neck, cervix, ovary, melanoma and many other tumor types[86–88]. Tumor cells that are sensitive to retinoic-acid-induced phenotypic changes (growth inhibition, differentiation or apoptosis) will exhibit a large increase in RAR-β2 expression when retinoic acid is administered[89,90]. In many instances, the reduced expression of RAR-β2 appears to result from increased DNA methylation in the promoter region of this gene[91]. Treatment with DNA demethylating agents, combined with retinoic acid, often leads to restoration of RAR-β2 expression, which is accompanied by growth inhibition[92,93].
Use of animal models to validate biomarkers
Based on the experience of the ATBC and CARET trials, where negative results or increased development of lung cancer were found[94,95], it is imperative that animal trials be used to identify useful chemopreventive agents, intermediate biomarkers and any potential harmful effects. Indeed, subsequent studies with ferrets given the high dose of beta carotene used in the clinical studies and exposed to smoke showed that these animals developed squamous metaplasia[96]. Valid animal models need to develop cancer at the appropriate organ site, with the tumor having the pathological and molecular signatures of the cognate human tumor. Very few carcinogen-induced or transgenic animals pass this stringent test.
The use of retinoids for chemoprevention in transgenic animal models of cancer has been limited. McCormick et al[97] found that 4-HPR given after N-ethyl-N-nitrosurea (ENU) administration to pim-1 oncogene overexpressing mice, delayed T-cell lymphoma development. However, intermediate biomarkers were not investigated in this study. The C3(1)SV40 large T/t-antigen (Tag) transgenic mouse has been developed to model human breast carcinogenesis. These mice develop mammary epithelial dysplasia that pro-gresses to mammary intraepithelial neoplasia, which is similar to ductal carcinoma found in situ in humans[98]. At 16 weeks these mice develop invasive carcinoma. Retinoic acid was found to inhibit mammary neoplasia in these mice when treatment was started at 5 weeks of age. A selective RXR analog, LGD1069, also inhibited tumor incidence and multiplicity in this transgenic model[99]. Using the Wistar-Unilever rat model of prostate cancer, McCormick and Rao[100] showed that 9-cis-retinoic acid was the most potent inhibitor of prostate carcinogenesis identified at that time (1999). A small number of other studies have shown chemoprevention effects of retinoids in animal models of bladder, pancreas and brain cancers[101–103]. None of these studies investigated intermediate biomarkers that could be used to predict the response of the tumor to retinoid treatment.
Summary, conclusions and future needs
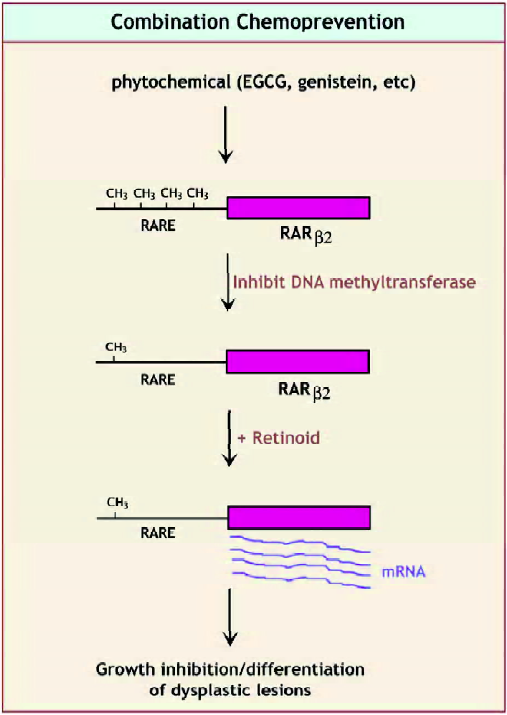
Retinoids have been shown to have chemopreventative activity for a number of tumors. The most studied are upper aerodigestive tract tumors. However, there are no well-defined and accepted intermediate biomarkers to validate the activity of retinoids in preventing tumor formation. Perhaps the marker that shows the most promise is RAR-β2. Its expression and/or inducibility by retinoic acid are decreased in most tumors that have been examined. Methylation of the RAR-β2 promoter has been documented in a number of retinoid-resistant tumors and may account for some of the silencing of this gene. However, because histone deacetylase inhibitors also increase the expression of RAR-β2, other epigenetic mechanisms are likely to be involved in the regulation of its expression. There are now a number of transgenic animal models that, at least partially, recapitulate the development of human cancer. Unfortunately, only a few of these new models have been tested for the ability of retinoids to inhibit tumor formation, and little research has been carried out to identify intermediate biomarkers in situations where retinoids have been shown to be effective. Future needs are to document RAR-β2 and other, perhaps tissue-specific, biomarkers for retinoid efficacy in chemoprevention. This could be accomplished most conveniently with transgenic animal models. In addition, the combination of retinoids (see Figure 3) with phytochemicals that act epigenetically to inhibit DNA methyltransferases or histone deacetylases on cancer chemoprevention is an area that deserves more intense investigation.
References
- Hofmann C, Eichele G. Retinoids in development. In: Sporn M, Roberts AB, Goodman M, editors. The retinoids, biology, chemistry and medicine, 2nd ed. New York: Plenum Press; 1994. p387–441.
- Eskild W, Hansson V. Vitamin A functions in the reproductive organs. In: R Blomhoff, editor. Vitamin A in health and disease. New York: Marcel Dekker; 1994. p531–9.
- DeLuca LM. Retinoids and their receptors in differentiation, embryogenesis and neoplasia. FASEB J 1991;5:2924-33.
- Wald G. The molecular basis of visual excitation. Nature 1968;219:800-07.
- Thompson JN, Howell JM, Pitt GAJ. Vitamin A and reproduction in rats. Proc R Soc Lond B 1964;159:510-35.
- Wolbach SB, Howe PR. Tissue changes following deprivation of fat-soluble A vitamin. Proc Soc Exp Biol Med 1925;77:825.
- Lasnitzki I. The influence of hypervitaminosis on the effect of 20-methylcholanthrene on mouse prostate glands growth in vitro. Br J Cancer 1955;9:434-41.
- Saffiotti U, Montessano R, Sellakumar AR, Borg SA. Experimental cancer of the lung: Inhibition by vitamin A of the induction of tracheobronchial squamous metaplasia and squamous cell tumors. Cancer 1967;20:857-64.
- Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007;315:820-5.
- Pasutto F., Stricht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, disphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet 2007;80:550-60.
- Levin MS, Li E, Ong DE, Gordon JI. Comparison of the tissue-specific expression and developmental regulation of two closely linked rodent genes encoding cytosolic retinol-binding proteins. J Biol Chem 1987;262:7118-25.
- Blaner WS, Das K, Mertz JR, Das SR, Goodman DS. Effects of dietary retinoic acid on cellular retinol- and retinoic acid-binding protein levels in various rat tissues. J Lipid Res 1986;27:1084-8.
- Astrom A, Tavakkol A, Pattersson U, Cromie M, Elder JT, Voorhees JJ. Molecular cloning of two human cellular retinoic acid-binding proteins (CRABP). J Biol Chem 1991;266:17662-6.
- Rajan N, Kidd GL, Tamage DA, Blaner WS, Suhara A, Godman DS. Cellular retinoic acid-binding protein messenger RNA levels in rat tissue and localization in rat testis. J Lipid Res 1991;32:1195-204.
- Ross AC, Zolfaghari R, Weisz J. Vitamin A: recent advances in the biotransformation, transport, and metabolism of retinoids. Curr Opin Gastroenterol 2001;17:184-92.
- Delva L, Bastie JN, Rochette-Egly C, Kraiba R, Balitrand N, Despouy G, et al. Physical and functional interactions between cellular retinoic acid binding protein II and the retinoic acid-dependent nuclear complex. Mol Cell Biol 1999;19:7158-67.
- Budh AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol 2002;22:2632-41.
- Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, et al. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992;68:397-406.
- Levin AA, Sturgenbeker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, et al. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXRα. Nature 1992;355:359-61.
- Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 1990;345:224-9.
- Predke PF, Zamble D, Sarkar B, Giguere V. Ordered binding of retinoic acid and retinoid-X receptors to asymmetric response elements involves determinants adjacent to the DNA-binding domain. Mol Endocrinol 1994;8:31-9.
- Carlsberg C, Bend I, Ways A, Meier E, Sturzenbecker LJ, Grippo JF, et al. Two nuclear signaling pathways for vitamin D. Nature 1993;361:657-60.
- Kliewer SA, Umensono K, Mangelsdorf DJ, Evans RM. Retinoid X receptors interact with nuclear receptors in retinoic acid, thyroid hormone, and vitamin D3 signaling. Nature 1992;335:446-9.
- Leid M, Kastner P, Lyons R, Nakshari H, Saunders M, Zacharewski T, et al. Purification, cloning, and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell 1992;68:377-95.
- Bugge TH, Pohl J, Lonnoy O, Stunnenberg HG. RXRγ, a promiscuous partner of retinoic acid and thyroid hormone receptors. EMBO J 1992;11:1409-10.
- Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferators-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA 1993;90:2160-4.
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferators signaling pathways through heterodimer formation of their receptors. Nature 1992;358:771-4.
- Kliewer SA, Umesono K, Heyman RH, Mangelsdorf DJ, Dyck JA, Evands RM. Retinoid X receptor–COUP-TF interactions modulate retinoic acid signaling. Proc Natl Acad Sci USA 1992;89:1448-52.
- Perlmann T, Jansson L. A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev 1995;9:769-82.
- Zhang XK, Hoffmann B, Tran PBV, Graupner G, Pfahl M. Retinoid X receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature 1992;355:441-6.
- Sucov HM, Murakami KK, Evans RM. Characterization of an autoregulated response element in the mouse retinoic acid receptor type β gene. Proc Natl Acad Sci USA 1990.5392-6.
- Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991;65:1255-66.
- Mader S, Leroy O, Chen JY, Chambon P. Multiple parameters control the selectivity of nuclear receptors for their response elements: selectivity and promiscuity in the response element recognition by retinoic acid receptors and retinoid X receptors. J Biol Chem 1993;268:591-600.
- Jho SH, Radoja N, Im MJ, Tomic-Canic M. Negative response elements in keratin genes mediate transcriptional repression and the cross-talk among nuclear receptors. J Biol Chem 2001;276:45914-20.
- Davies RE. Effect of vitamin A on 7,12-dimethylbenz(a)anthracene-induced papillomas in rhino mouse skin. Cancer Res 1967;27:237-41.
- Harris CC, Sporn MB, Kaufman DG, Smith JM, Jackson FE, Saffiotti U. Histogenesis of squamous metaplasia in the hamster tracheal epithelium caused by vitamin A deficiency of benzo[a]pyrene Ferric oxide. J Natl Cancer Inst 1972;48:743-61.
- Rogers AE, Herndon BJ, Newberne PM. Induction by dimethylhydrzine of intestinal carcinoma in normal rats and rats fed high or low levels of vitamin A. Cancer Res 1973;33:1003-9.
- McCormick DL, Burns FJ, Albert RE. Inhibition of benz[a]pyrene-induced mammary carcinogenesis by retinyl acetate. J Natl Cancer Inst 1981;66:559-64.
- Nauss KM, Bueche D, Newberne PM. Effect of vitamin A nutriture on experimental esophageal carcinogenesis. J Natl Cancer Inst 1987;79:145-7.
- DeLuca LM, Tarone R, Huynh M, Jones CS, Chen LC. Dietary retinoic acid inhibits mouse skin carcinogenesis irrespective of age of initiation. Nutr Cancer 1996;25:249-57.
- Hankin JH, Kolonel LN, Hinds MW. Dietary history methods for epidemiologic studies: application in a case-control study of vitamin A and lung cancer. J Natl Cancer Inst 1984;73:1417-22.
- Mettlin CJ. Epidemiology of vitamin A and aerodigestive cancer. Adv Exp Med Biol 1992;320:21-6.
- Moon RC. Vitamin A, retinoids and breast cancer. Adv Exp Med Biol 1994;364:101-07.
- Guo Z, Nanus DM, Ruiz A, Rando RR, Bok D, Gudas LJ. Reduced levels of retinyl esters and vitamin A in human renal cancers. Cancer Res 2001;61:2774-81.
- Guo Z, Knudsen BS, Peehl DM, Ruiz A, Bok D, Rando RR, et al. Retinol metabolism and lecithin:retinol acyltransferase levels are reduced in cultured human prostate cancer cells and tissue specimens. Cancer Res 2002;62:1654-61.
- Jeronimo C, Oliveria HR, Lobo F, Pais I, Teixeira MR, Lopes C. Aberrant cellular retinol binding protein 1 (CRBP1) gene expression and promoter methylation in prostate cancer. J Clin Pathol 2004;57:872-6.
- Van Wauwe JP, Coene MC, Goossens J, Van Nijen G, Cools W, Lauwers W. Ketoconazole inhibits the in vitro and in vivo metabolism of all-trans-retinoic acid. J Pharmacol Exp Ther 1988;245:718-22.
- Soprano DR, Qin P, Soprano KF. Retinoic acid receptors and cancers. Ann Rev Nutr 2004;24:201-21.
- McGregor F, Wagner E, Felix D, Soutar D, Parkinson K, Harrison PR. Inappropriate retinoic acid receptor β expression in oral dysplasias: correlation with acquisition of the immortal phenotype. Cancer Res 1977;57:3886-9.
- Picard E, Seguin C, Momhoven N, Rochette-Egly C, Siat J, Borrelly J, et al. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J Natl Cancer Inst 1999;91:1059-66.
- Qiu H, Zhang W, El-Naggar AK, Lippman SM, Lin P, Lotan R, et al. Loss of retinoic acid receptor β expression is an early event during esophageal carcinogenesis. Am J Pathol 1999;155:1519-23.
- Swisshelm K, Ryan K, Lee X, Tsou HC, Peacocke M, Sager R. Down-regulation of retinoic acid receptor β in mammary carcinoma cell lines and its up-regulation in senescing normal mammary epithelial cells. Cell Growth Differ 1994;5:133-41.
- Zhang XK, Liu Y, Lee MO, Pfahl M. A specific defect in the retinoic acid response associated with human lung cancer cell lines. Cancer Res 1994;54:5663-9.
- Lin B, Chen G, Xiao D, Lollure SK, Cao X, Su H, et al. Orphan receptor COUP-TF is required for induction of retinoic acid receptor, growth inhibition and apoptosis by retinoic acid in cancer cells. Mol Cell Biol 2000;20:957-70.
- Wu Q, Li Y, Liu R, Agadir A, Lee MO, Liu Y, et al. Modulation of retinoic acid sensitivity in lung cancer cells through dynamic balance of orphan receptors nurr77 and COUP-TF and their heterodimerization. EMBO J 1997;16:1545-69.
- Arapshian A, Kuppumbatti YS, Mira-y-Lopez R. Modulation of conserved CpG sites neighboring the beta retinoic acid response element may mediate retinoic acid receptor β gene silencing in MCF7 breast cancer cells. Oncogene 2000;19:4066-70.
- Cote S, Sinnett D, Momparler RL. Demethylation by 5-aza-2’ deoxycytidine of specific 5-methylcytosine sites in the promoter region of the retinoic acid receptor β gene in human colon carcinoma cells. Anticancer Drugs 1998;9:743-50.
- Nakayama T, Watanabe M, Yamanaka M, Hirokawa Y, Suzuki H, Ito H, et al. The role of epigenetic modifications in retinoic acid receptor β2 gene expression in human prostate cancers. Lab Invest 2001;81:1049-57.
- Cote S, Momparier RL. Activation of the retinoic acid receptor β gene by 5-aza-2’-deoxycytidine in human DLD-1 colon carcinoma cells. Anticancer Drugs 1995;8:56-61.
- Sirchia SM, Ren M, Pili R, Siron E, Somenzi G, Ghidoni R, et al. Endogenous reactivation of the RARβ2 tumor suppressor gene epigenetically silenced in breast cancer. Cancer Res 2002;62:2455-61.
- Hong WK, Endicott J, Itri LM, Doos W, Batsakis JG, Bell R, et al. 13-cis-retinoic acid in the treatment of oral leukoplakia. N Engl J Med 1986;315:1501-5.
- Lippman SM, Batsakis JG, Toth BB, Weber RS, Lee JJ, Martin JW, et al. Comparison of low-dose isotretinoin with beta carotene to prevent oral carcinogenesis. N Engl J Med 1993;328:15-20.
- Stich HF, Hornby AP, Mathew B, Sankaranarayanan R, Nair MK. Response of oral leukoplakias to the administration of vitamin A. Cancer Lett 1988;40:93-101.
- Han J, Jiao L, Sun Z, Gu QM, Scanlon KJ. Evaluation of N-4-(hydroxycarbophenyl) retinamide as a cancer prevention agent and as a cancer chemotherapeutic agent. In Vivo 1990;4:153-60.
- Costa A, Formelli F, Chiesa F, Decensi A, de Paol G, Veronesi U. Prospects of chemoprevention of human cancers with the synthetic retinoid fenretinide. Cancer Res 1994;54:2032s-7s.
- Lotan R, Xu XC, Lippman SM, Ro JY, Lee JS, Lee JJ, et al. Suppression of retinoic acid receptor β in premalignant oral lesions and its up-regulation by isotretinoin. N Engl J Med 1995;332:1405-10.
- Misset JL, Mathe G, Santelli G, Gouveia J, Homasson JP, Sudre MC, et al. Regression of bronchial epidermoid metaplasia in heavy smokers with etretinate treatment. Cancer Detect Prev 1986;9:167-70.
- Lippman SM, Kessler JF, Meyskens FL. Retinoids as preventive and therapeutic anticancer agents. Cancer Treat Rep 1987;71:391-405.
- Kligman AM, Thorne EG. Topical therapy of actinic keratosis with tretinoin. In: Marks R, editor. Retinoids in cutaneous malignancy. Cambridge: Blackwell Scientific; 1991. p66–73.
- Moriarty M, Dunn J, Darragh A, Lambe R, Brick I. Etretinate in treatment of actinic keratosis: A double blind crossover study. Lancet 1982;1:364-5.
- Watson AB. Preventive effect of etretinate therapy on multiple actinic keratoses. Cancer Detect Prev 1986;9:161-5.
- Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 1988;318:1633-7.
- Kraemer K, Di Giovanna JJ, Peck GL. Oral isotreinoin prevention of skin cancer in xeroderma pigmentosum: Individual variation in dose response. J Invest Dermatiol 1990;94:544.
- Kelly JW, Sabto J, Gurr FW. Retinoids to prevent skin cancer in organ transplant recipients. Lancet 1991;338:1407.
- Moon TE, Levine N, Cartmel B, Bangert JL. Retinoids in prevention of skin cancer. Cancer Lett 1997;114:203-5.
- Greenberg ER, Baron JA, Stukel TA, Stevens MM, Mandel JS, Spencer SK. A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. N Engl J Med 1990;323:789-95.
- Tangrea JA, Edwards BK, Taylor PR, Hartman AM, Peck GL, Salasche SJ, et al. Long term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: A multicenter clinical trail. J Natl Cancer Inst 1992;84:328-32.
- Graham V, Surwit ES, Weiner S, Meyskens FL. Phase II trial of beta-all-trans-retinoic acid for cervical intraepithelial neoplasia delivered via a collagen sponge and cervical cap. West J Med 1986;145:192-5.
- Meyskens FL, Surwit E, Moon TE, Childers JM, Davis JR, Dorr RT, et al. Enhancement of regression of cervical intraepithelial neoplasia II (moderate dysplasia) with topically applied all-trans-retinoic acid: a randomized trial. J Natl Cancer Inst 1994;86:539-43.
- Lawrence JA, Merino MJ, Simpson JF, Manrow RE, Page DL, Steeg PS. A high risk lesion for invasive breast cancer, ductal carcinoma in situ, exhibits frequent overexpression of retinoid X receptor. Cancer Epid Biomarkers Prevent 1998;7:29-35.
- Elmore FL, Jain A, Siddiqui S, Tohidian N, Meyskens FL, Steele VE, et al. Development and characteristics of a human cell assay for screening agents for melanoma prevention. Melanoma Res 2005;17:42-50.
- Papadimitrakopoulou VA, Hong WK. Biomolecular markers as intermediate end points in chemoprevention trials of upper aerodigestive tract cancer. Int J Cancer 2000;88:852-5.
- Lippman SM, Shin DM, Lee JJ, Batsakis JG, Lotan R, Tainsky MA, et al. p53 and retinoid chemoprevention of oral carcino-genesis. Cancer Res 1995;55:16-19.
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999;96:8681-6.
- Xu XC, Lee JS, Lee JJ, Morice RC, Liu X, Lippman SM. Nuclear retinoid receptor beta in bronchial epithelium of smokers before and during chemoprevention. J Natl Cancer Inst 1999;91:1317-21.
- Widschwendter M, Berger J, Daxenbichler G, Muller-Holzner E, Widschwendter A, Mayr A, et al. Loss of retinoic acid receptor expression in breast cancer and morphologically normal adjacent tissues but not in the normal breast tissue distant from the cancer. Cancer Res 1997;57:4158-61.
- Picard E, Seguin C, Momhoven N, Rochette-Egly C, Siat J, Borrelly J, et al. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J Natl Cancer Inst 1999;91:1059-66.
- Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004;23:4014-22.
- Seewaldt VL, Johnson BS, Parker MB, Collins SJ, Swisshelm K. Expression of retinoic acid β mediates retinoic acid-induced growth arrest and apoptosis in breast cancer cells. Cell Growth Differ 1995;6:1077-88.
- Xu XC, Liu X, Tahara E, Lippman SM, Lotan R. Expression and up-regulation of retinoic acid receptor-beta is associated with retinoid sensitivity and colony formation in esophageal cancer cell lines. Cancer Res 1999;59:2477-83.
- Hayashi K, Yokozaki H, Goodison S, Oue N, Suzuki T, Lotan R, et al. Inactivation of retinoic acid receptor β by promoter CpG hypermethylation in gastric cancer. Differentiation 2001;69:13-21.
- Yang Q, Shan L, Yoshimura G, Nakamura M, Nakamura Y, Suzuma T, et al. 5-aza-2’-deoxycytidine induces retinoic acid receptor beta2 demethylation, cell cycle arrest and growth inhibition in breast carcinoma cells. Anticancer Res 2002;22:2753-6.
- Mongan NP, Gudas LJ. Valproic acid, in combination with all-trans retinoic acid and 5-aza-2’-deoxycytidine, restores expression of RARβ2 in breast cancer cells. Mol Cancer Ther 2005;4:477-86.
- Albanes D, Heinonen OP, Huttunen JK, Taylor PP, Virtamo J, Edwards BK, et al. Effects of alpha-tocopherol and beta-carotene supplements on cancer incidence in the Alpha Tocopherol Beta-carotene Cancer Prevention Study. Am J Clin Nutr 1995;62:1425S-30S.
- Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst 1966;88:1550-9.
- Wang XD, Liu C, Bronson RT, Smith DE, Krinsky NI, Russell M. Retinoid signaling and activator protein-1 expression in ferrets given β-carotene supplements and exposed to tobacco smoke. J Natl Cancer Inst 1999;91:60-6.
- McCormick DL, Johnsen WD, Rao KV, Bowman-Gram T, Steele VE, Lubet RA, et al. Comparative activity of N-(4-hydroxy-phenyl)-all-trans-retinamide and α-difluoromethylornithine as inhibitors of lymphoma induction in PIM transgenic mice. Carcinogenesis 1996;17:2513-7.
- Wu K, Kim HT, Rodiquez JL, Munoz-Medellin D, Mohsin SK, Hilsenbeck SG, et al. 9-cis Retinoic acid suppresses mammary tumorigenesis in C3(1)-simian virus 40 T antigen-transgenic mice. Clin Cancer Res 2000;6:3696-704.
- Wu K, Kim HT, Rodriquez JL, Hilsenbeck SG, Mohsin SK, Xu XC, et al. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol Biomarkers Prev 2002;11:467-74.
- McCormick DL, Rao KV. Chemoprevention of hormone-dependent prostate cancer in the Wistar–Unilever rat. Eur Urol 1999;35:464-7.
- Becci PJ, Thompson HJ, Strum JM, Brown CC, Sporn MB, Moon RC. N-butyl-N-(4-hydroxybutyl)nitrosamine-induced bladder cancer in C57BL/6 X DBA/2F1 mice as a useful model for study of chemoprevention of cancer with retinoids. Cancer Res 1981;41:927-32.
- Curphey TJ, Kuhlmann ET, Roebuck BD, Longnecker DS. Inhibition of pancreatic and liver carcinogenesis in rats by retinoid- and selenium-supplemented diets. Pancreas 1988;3:36-40.
- Ross DA, Kish P, Muraszko KM, Blaivas M, Srawderman M. Effect of dietary vitamin A or N-acetylcysteine on ethylnitrosurea-induced rat gliomas. J Neurooncol 1998;40:29-38.