
Vitamin D and prevention of breast cancer1
Vitamin D and cancer prevention
Although originally identified based on its ability to prevent the bone disease rickets, it is now recognized that 1a,25 dihydroxyvitamin D3 (1,25D), the biologically active form of vitamin D3, exerts effects in almost every tissue in the body. Recent epidemiological studies have focused attention on a possible link between vitamin D and cancer. The concept underlying this link is fairly simple: that the vitamin D receptor (VDR) and its ligand 1,25D induce a program of gene expression that contributes to maintenance of the quiescent, differentiated phenotype. This concept predicts that the vitamin D system might have relevance for both prevention and treatment of cancer. The initial identification of the VDR in breast cancer cells suggested that this receptor might represent a target for breast cancer therapy, and multiple studies have assessed the effects of vitamin D on breast cancer cells in vitro and tumors in vivo. In addition, the potential side effects of vitamin D therapy have been characterized, and a combination of trials of vitamin D compounds with standard therapies such as anti-estrogens, chemotherapeutic drugs and radiation have been reported. For the most part, these studies have been limited to animal models of breast cancer such as the NMU-induced breast cancer model in rats and human xenograft models in immunosuppressed mice.
Studies to test the role of vitamin D in breast cancer prevention have included characterization of the expression and function of the vitamin D pathway in normal mammary tissue using animal models and human cells. Studies with the VDR knockout mouse model have been highly informative in determining whether complete abrogation of vitamin D signaling alters the susceptibility of mammary tissue to cancer development. In this review, we summarize the currently available data generated from both in vitro and in vivo studies, with an emphasis on the cellular and molecular mechanisms by which vitamin D may contribute to breast cancer prevention.
Overview of vitamin D biology
The nutritional substance termed “vitamin D” comprises ergocalciferol (vitamin D2, from plant sources) and cholecalciferol (vitamin D3, from animal sources). Both forms can be obtained from the diet, but very few natural foods have significant vitamin D activity[1]. In many countries, including the US, fortification of foods such as milk and orange juice with vitamin D3 is common. Cholecalciferol can also be generated from 7-dehydrocholesterol in the epidermis upon exposure to ultraviolet (UV) radiation, thus, vitamin D is not technically a vitamin. In fact, because the concentration of vitamin D in natural foods is quite low, the majority of vitamin D in most individuals likely originates from epidermal synthesis, however there is considerable individual variation in this process[2]. In particular, epidermal synthesis is affected by skin pigmentation, sunscreen use, age, season, latitude and other lifestyle factors. Despite the fortification of vitamin D3 in foods and endogenous synthesis, the prevalence of vitamin D insufficiency is surprisingly common, especially in populations living in northern climates and in the elderly[3-6]. Particularly relevant to the possible relationship between vitamin D and breast cancer, vitamin D deficiency has been reported in a high percentage of women, including during adolescence, pregnancy and/or lactation and after menopause, even in sunny climates[3,7-9]. These and other determinants of vitamin D status in humans are summarized in Table 1.

Full table
The increasing number of reports of vitamin D insufficiency has prompted re-evaluation of the recommended adequate intake for vitamin D[5,10] which was directed against prevention of rickets. There is fairly compelling evidence that prolonged sub-clinical vitamin D deficiency, which may not be associated with hypocalcemia or bone disease but could limit availability of active vitamin D metabolites to tissues, contributes to chronic disease in human populations. However, relevant biomarkers of vitamin D status that reflect newly identified actions in colon, prostate and breast that are relevant for cancer prevention (as discussed below) remain to be identified.
Both naturally occurring forms of vitamin D require metabolism for biological activity, and defects in activation pathways can affect vitamin D function[11]. For simplicity, this review focuses on vitamin D3, but the metabolism and function of vitamin D2 is similar, although some data suggest that D2 is less potent than D3 in humans. Regardless of source (endogenous synthesis or diet), the initial step in metabolism of vitamin D3 is hydroxylation at the 25 position, generating 25-hydroxyvitamin D3 (25D). The 25D metabolite, which is mainly produced in the liver, is the major circulating form and the most accurate biomarker of overall vitamin D3 status[6,12]. Further metabolism of 25D occurs in many tissues and leads to generation of multiple metabolites, two of these -24,25-dihydroxyvitamin D3 (24,25D) and 1α,25-dihydroxyvitamin D3 (1,25D), have been extensively characterized in relation to maintenance of calcium homeostasis. Generation of 24,25D is catalyzed by 24-hydroxylase (also termed CYP24A1), an enzyme present in most vitamin D target tissues. The 24,25D metabolite does not avidly bind VDR, and its production is considered the first step in degradation of 25D. Production of 1,25D, the biologically active metabolite, is mediated by 1α-hydroxylase (also termed CYP27B1), an enzyme that is highly expressed in renal proximal tubules. The importance of renal 1α-hydroxylase in maintaining systemic 1,25D has been documented in patients with end stage renal failure, who require exogenous supplementation with this metabolite to prevent renal osteodystropy. Although originally thought to be exclusively localized to the kidney, 1α-hydroxylase gene expression and activity has now been localized to multiple other tissues including mammary gland[13-16]. In extra-renal cell types, 1α-hydroxylase likely generates 1,25D, which acts locally within tissues, as circulating 1,25D is virtually undetectable in anephric individuals. The 1,25D metabolite, sometimes called calcitriol, is the high affinity ligand for the VDR, a nuclear transcription factor (Figure 1).
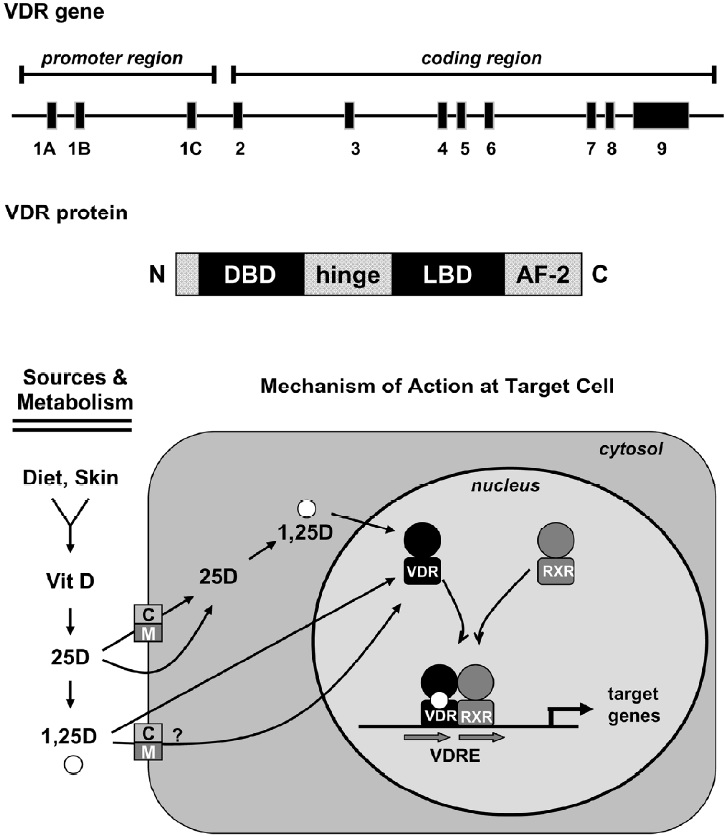
Vitamin D: cellular uptake and general mecha-nism of action
Vitamin D metabolites, including 25D and 1,25D, circulate as free steroids and in complex with the vitamin D binding protein (DBP), a member of the albumin gene family. The 1,25D metabolite is presumed to enter cells via diffusion through the plasma membrane, however, uptake of this metabolite via active processes, either as the free steroid or in complex with DBP, has not been ruled out. The 25D meta-bolite binds to DBP with 20–30-fold higher affinity than does 1,25D, and the 25D-DBP complex has been shown to enter renal cells via receptor-mediated endocytosis. This process is facilitated by the megalin-cubilin endocytic receptor complex present on the renal cell plasma membrane, which is essential for maintenance of vitamin D homeostasis in vivo[17]. However, the relative contribution of facilitated versus passive uptake mechanisms for physiologically significant vitamin D steroids in different cell types has yet to be thoroughly characterized, either in vitro or in vivo.
Once internalized, the metabolic fate of 25D reflects its trafficking to metabolizing enzymes and the relative expression and/or activity of the 24- and 1-hydroxylases. In cells with high levels of 24-hydroxylase, generation of 24,25D and subsequent catabolism would predominate, precluding VDR activation. However, cells with functional 1α-hydroxylase could potentially convert 25D to 1,25D, which could bind to VDR and mediate tissue-specific cell regulatory effects in an autocrine fashion. The implication of the autocrine pathway is that local cellular production of 1,25D would likely be regulated in a tissue-specific manner independently from systemic calcium homeostasis. Similarly, the actions of locally produced 1,25D would be confined to the immediate cellular environment and would not necessarily affect body calcium homeostasis. Existence of the autocrine pathway also implies that circulating 25D becomes the critical determinant of cellular vitamin D activity and necessitates re-definition of the optimal serum levels of 25D needed for the maintenance of local 1,25D generation.
Whether generated in cells from 25D or taken up from the circulation, 1,25D binds to the VDR, a member of the steroid receptor family of ligand-dependent transcription factors that modulate gene expression in a tissue-specific manner[18]. In an early study, 23 of 33 established human cancer cell lines surveyed expressed VDR[19]. Expression profiling of breast, prostate, and squamous carcinoma cells has identified 1,25D responsive gene clusters involved in the regulation of cell cycle, differentiation, cell adhesion and immune responses, indicating a diverse and broad range of VDR targets potentially involved in cell regulation[20-23]. Like other nuclear receptors, gene regulation by the liganded VDR requires dimerization, most often with the retinoid X receptor (RXR) family, and binding to specific DNA sequences in target gene promoters[24]. Although a variety of structurally distinct vitamin D responsive elements have been identified, the best characterized is a hexanucleotide direct repeat separated by three variable base pairs (DR3) to which VDR:RXR hetero-dimers bind[25]. Additional mechanisms of genomic VDR signaling include interacting with partners other than RXR, binding to diverse DNA sequences, and ligand independent effects[26,27]. VDR can also influence gene expression via interactions with other transcription factors such as Sp1[28]. In addition, the VDR is subject to post-translational modifica-tions, including phosphorylation, that affect its transcriptional activity[29,30].
In addition to genomic signaling, 1,25D can exert rapid effects on signal transduction pathways, leading to biological responses at the plasma membrane or in the cytoplasm[18]. Identification of an alternative binding pocket in the VDR for ligands that mediate rapid effects suggests that the VDR mediates some of these non-genomic effects, a suggestion supported by studies with cells from VDR null mice[31,32]. Localization of the nuclear VDR protein to caveolae, specialized signaling complexes present in plasma membrane, further supports this concept[33]. Examples of non-transcriptional effects of the 1,25D – VDR complex with potential relevance to cancer cell regulation include regulation of ion channels, protein kinase C activation, interaction with β-catenin and activation of protein phosphatases PP1c and PP2Ac[34-36]. The possibility that alternative receptors for vitamin D metabolites that have been linked to rapid responses[37] may contribute to cancer cell regulation by 1,25D has yet to be thoroughly investigated. Thus, the relative contributions of genomic and non-genomic signaling in mediating the diverse biological effects of 1,25D, particularly in relation to its anti-cancer properties, remain to be fully clarified.
Effects of vitamin D on breast cancer cells and tumors
In response to the initial identification of VDR in cancer cells, numerous studies examined the effects of 1,25D on transformed cells[38]. Furthermore, a large number of structural analogs of vitamin D developed by pharmaceutical companies and academic researchers have been used to probe the mechanisms of vitamin D-mediated growth inhibition[39-41]. In the following sections, data generated with both natural vitamin D metabolites and synthetic analogs is discussed, and readers are referred to the original citations for details.
In both estrogen receptor positive and negative breast cancer cells, 1,25D induces cell cycle arrest, differentiation and apoptosis [13,42-46]. The anti-proliferative effects of 1,25D result from alterations in key cell cycle regulators thatculmin-ate in de-phosphorylation of the retinoblastoma protein and arrest of cells in G0/G1[46] . The cyclin dependent kinase inhibitors p21 and/or p27 are genomic targets of the 1,25D- VDR complex in many cell types [28,47,48]. In addition to direct regulation of cell cycle modulators, 1,25D blocks mitogenic signaling, including that of estrogen, EGF, IGF-1 and KGF and upregulates negative growth factors such as TGF-β[49-52].
In some transformed cells, 1,25D induces apoptotic cell death via generation of reactive oxygen species, dissipation of the mitochondrial membrane potential and cytochrome c release[43,53], features of the intrinsic (mitochondrial) pathway of apoptosis. Furthermore, 1,25D exerts additive or synergistic effects in combination with other triggers of apopto-sis, such as radiation and chemotherapeutic agents[54-57]. In MCF-7 cells, 1,25D down regulates the anti-apoptotic protein Bcl-2 and induces the redistribution of the pro-apoptotic protein Bax from cytosol to mitochondria[43,53]. Furthermore, overexpression of Bcl-2 renders breast cancer cells resistant to 1,25D-mediated apoptosis[53]. The role of caspases and other proteases in 1,25D-mediated cell death appears to vary with cell type. Activation of caspases 3 and 9 occurs during 1,25D-induced apoptosis in some cells, and caspase inhibition can prevent some features of 1,25D-mediated apoptosis, however, caspase inhibitors do not prevent 1,25D-mediated death[43,58]. Other proteases implicated in 1,25D-mediated cell death include calpains and cathepsins[58-61]. Collectively, these studies indicate that a wide variety of different signaling pathways, apoptotic regulatory proteins and proteases may contribute to 1,25D-mediated apoptosis depending on the specific cell type and/or context.
Although cells lacking functional p53 retain sensitivity to 1,25D[53], the VDR has been identified as a transcriptional target of p53 and the related proteins p63 and p73[62,63]. These studies suggest that VDR regulated pathways may contribute to the tumor suppressive effects of the p53 family. In support of this notion, VDR and p53 mediate similar biological effects (growth arrest in G0/G1, apoptosis, DNA repair) via common target genes (p21, bax, GADD45). On the p21 promoter, both independent and overlapping VDR and p53 binding sites have been characterized[64].
To examine whether the anti-cancer effects of 1,25D are mediated by the nuclear VDR, we developed mammary tumor cell lines from VDRKO mice[65]. Tumors were induced in mice lacking VDR and their normal wild type (WT) littermates, and cell lines were established from these tumors. WT cell lines expressed VDR and underwent growth inhibition and apoptosis when treated with1,25D, whereas VDRKO cell lines did not express VDR and did not exhibit growth arrest or apoptosis when treated with 1,25D or synthetic vitamin D analogs. Interestingly, cells lacking VDR retained sensitivity to retinoids, anti-estrogens and DNA damaging agents such as etoposide. These data indicate that VDR specifically mediates the growth inhibitory effects of vitamin D steroids, but is not absolutely required for the anti-cancer effects of unrelated agents. Given the known interactions between VDR and p53, however, subtle differences in the sensitivity of VDRKO cells to diverse apoptotic agents have not been ruled out. More detailed studies are clearly needed to determine whether 1,25D and the VDR exert independent effects on pathways involved in DNA damage sensing and repair, cell cycle regulation or apoptosis.
Although therapeutic use of 1,25D is precluded by dose-limiting calcemic toxicity, synthetic analogs of 1,25D that exhibit less potent calcemic effects have provided proof of the principle that VDR agonists can inhibit growth and induce regression of mammary tumors in animal models[66-68]. Furthermore, studies on xenografts derived from WT and VDRKO cells indicated that the expression of functional VDR in tumor epithelial cells (rather than in accessory cells such as fibroblasts, immune cells or endothelial cells) is necessary for the anti-tumor effects of vitamin D analogs in vivo (Valrance et al, in press). These studies definitively establish that the VDR is the mediator of the negative growth regulatory effects of vitamin D steroids in vivo.
Evidence for breast cancer prevention by vita-min D
In contrast to the extensive work demonstrating the effects of vitamin D on transformed mammary cells, there has been less emphasis on defining the role of vitamin D in breast cancer prevention. As noted above, the link between vitamin D and breast cancer prevention is based on the concept that 1,25D promotes or maintains the differentiated phenotype in normal mammary cells. Consistent with this concept, the VDR is expressed in normal mammary epithelial tissue in vivo and in non-transformed human mammary epithelial (HME) cells in vitro[13,69,70]. In mouse mammary gland, VDR is localized predominantly in differentiated epithelial cells, and its expression increases 100-fold during the course of pregnancy and lactation[70,71].
The function of the vitamin D pathway in HME cells has recently been evaluated. The effects of 1,25D on HME cells include growth arrest and induction of differentiation markers such as E-cadherin, but apoptosis has not been reported[13]. As in breast cancer cells, supra-physiological concentrations of 1,25D are required to elicit these effects. Notably, non-transformed mammary cells express CYP27B1, the enzyme that converts 25D to 1,25D, suggesting the possibility that 25D may be the biologically relevant metabolite in the mammary gland[13,14]. Indeed, mammary cells can bioactivate 25D to 1,25D and physiological concentrations of 25D can inhibit growth of HME cells in vitro[13,14]. A caveat to these studies is that very little is known about the delivery of 25D to mammary cells. As discussed earlier, 25D binds avidly to serum DBP, therefore it is likely that 25D is delivered to the mammary gland in complex with DBP. However, whether 25D dissociates from the 25D-DBP complex or whether the 25-DBP complex is internalized intact by mammary cells is unclear. Recent studies have demonstrated that both murine and human mammary epithelial cells express megalin and cubilin, proteins required for the endocytic uptake of DBP in kidney. Furthermore, uptake of DBP occurred in mammary cells in vitro and was correlated with 25D-mediated transactivation of VDR[72]. However, further studies are necessary to determine whether endocytosis of the 25D-DBP complex occurs in mammary tissue in vivo.
Animal studies also support the concept that vitamin D signaling reduces breast cancer development. Rodents fed western style diets (low in vitamin D and calcium, high in saturated fat) developed hyperproliferation and/or enhanced rates of tumor formation in colon, prostate and mammary gland compared to rats fed adequate calcium and vitamin D[73]. In mammary gland organ culture, 1,25D inhibited hormone-driven proliferation and reduced the number of carcinogen initiated pre-neoplastic lesions during both the initiation and the promotion stages, indicating that vitamin D signaling can exert direct anti-neoplastic effects at multiple steps in the carcinogenesis process[74]. VDR agonists have also been shown to inhibit angiogenesis, invasion and metastasis indicating a potential benefit of vitamin D on later stages of cancer progression[66,75,76].
Data from VDR null mice support a role for vita-min D in cancer prevention
Mice lacking the VDR demonstrate excess proliferation and branching as well as impaired apoptosis during the reproductive cycle compared to their normal counterparts[70,71]. Comparison of gene expression in normal and VDR knockout mice has identified cyclin D1, p21, clusterin, β-catenin and TGF-β1 as potential VDR target genes in the mammary gland in vivo (Zinser et al. unpublished data). Demonstration that VDR ablation alters growth regulatory pathways in mammary gland raised the possibility that VDRKO mice might display an enhanced risk for cancer development in this tissue. Indeed, the incidence of mammary hyperplasias and the development of ER negative tumors in response to the carcinogen DMBA was higher in VDRKO mice than their WT counterparts[77]. Furthermore, on the MMTV-neu transgenic background, VDR heterozygote mice demonstrated a higher incidence of neu-driven mammary tumors than did WT mice[78]. Notably, differences in cancer susceptibility were not limited to the mammary gland, as VDRKO mice displayed increased sensitivity to tumors in the lymph nodes and skin in response to DMBA compared to WT mice[77,79]. These in vivo studies have provided the most direct evidence that VDR signaling can protect against cancer development. Collectively, these and other animal studies have confirmed that the effects of vitamin D signaling observed in vitro translate to effects on cell proliferation, differentiation and apoptosis in vivo that are of sufficient magnitude to affect the carcinogenic process.
Effect of transformation on the vitamin D pathway
Some transformed breast cells display limited sensitivity to 1,25D, suggesting that the vitamin D pathway may be deregulated during cancer development. Multiple mechanisms have been identified that contribute to 1,25D resistance, including loss of VDR expression, alterations in transcriptional co-regulators and overexpression of CYP24, the enzyme that catabolizes 1,25D. Overexpression of the anti-apoptotic protein bcl-2 renders cancer cells resistant to 1,25D-mediated apoptosis and expression of certain oncogenes (including ras and SV40 large T antigen) interferes with vitamin D signaling[53,80,81]. Breast cancer cell lines have been selected for resistance to 1,25D in vitro[82,83]. These cell lines retain expression of VDR but exhibit changes in protein expression that favor autonomous growth signaling and downregulate the apoptotic pathway[84,85]. Amplification of the CYP24 gene was reported in human breast cancer and higher CYP24 expression was detected in tumors compared to adjacent normal tissue[14,86]. De-sensitization of breast cancer cells to growth inhibition by VDR ligands has also been associated with changes in nuclear receptor co-repressors via epigenetic mechanisms, which are potentially reversible[87,88]. These data indicate that cancer cells use multiple mechanisms to evade the negative growth regulatory effects of the vitamin D signaling pathway.
Vitamin D and breast cancer links at the population level
An evaluation of the Nurses Health Study found that intake of dairy products, dairy calcium and vitamin D were inversely associated with breast cancer risk in premenopausal, but not postmenopausal, women[89]. John et al[90] demonstrated that sunlight exposure and dietary vitamin D were associated with reduced risk of breast cancer, however, the association was dependent on the region of residence. A prospective analysis of breast cancer incidence in relation to vitamin D intake for over 30 000 participants in the Women’s Health Study indicated that higher intake of vitamin D was moderately associated with a lower risk of pre- but not post- menopausal breast cancer[91]. These data are consistent with reports of inverse association between vitamin D status and mammographic density in pre-menopausal women[92,93]. Correlation between exposure to solar radiation and breast cancer risk has also been suggested in large epidemiological studies[94,95]. A pooled analysis of studies that assessed serum 25D in relation to breast cancer demonstrated a clear dose-response relationship, with the highest quintile of serum 25D associated with a 50% reduction in breast cancer risk[96]. These data suggested that serum 25D concentrations above 50 ng/mL may be required to optimize vitamin D signaling in mammary tissue. However, studies have demonstrated that it is difficult to maintain serum 25D in this range from dietary sources, particularly when sunlight exposure is limited[97,98]. Furthermore, the amount of vitamin D present in currently available over the counter supplements (400 IU) is too low to significantly elevate serum 25D[5]. Collectively, these observations emphasize the need for re-evaluation of public health recommendations regarding sun exposure, vitamin D intake, food fortification and supplement use in relation to vitamin D status and chronic disease.
Conclusions
Table 2 summarizes the current evidence linking the vitamin D signaling pathway with breast cancer prevention. VDR is expressed in normal mammary cells, where it regulates proliferation, apoptosis and differentiation via distinct targets at different stages of development. In mice, deficiency of the VDR enhances risk for transformation in mammary gland, lymphoid tissue, skin and colon. The VDR ligand 1,25D inhibits growth and induces apoptosis in breast cancer cells and tumors, and these effects absolutely require the VDR. VDR ligands also inhibit growth of normal human mammary epithelial cells, and evidence suggests that autocrine bio-activation of vitamin D precursors can occur within mammary cells. Thus, data from both human tissues and animal models support the concept that the VDR and its ligand induce a program of gene expression that contributes to maintenance of the differentiated phenotype in breast cells, a concept that is consistent with a role for vitamin D in both prevention and treatment of breast cancer. At the current time, however, the amount of vitamin D (either from diet or endogenous synthesis) needed to optimize growth inhibitory signaling through the VDR in vivo is currently undefined, and further studies are needed before guidelines or requirements for human populations can be established. Collectively, these studies emphasize that multiple components of the vitamin D signaling system are present in normal mammary cells and emphasize the need for additional research on expression and function of these proteins in intact mammary tissue in vivo, particularly in relation to maintenance of the differentiated phenotype.

Full table
References
- Holick MF, Vitamin D. A millenium perspective. J Cell Biochem 2003;88:296-307.
- Binkley N, Novotny R, Krueger D, Kawahara T, Daida YG, Lensmeyer G, et al. Low Vitamin D Status Despite Abundant Sun Exposure. J Clin Endocrinol Metab 2007. in press.
- Gordon CM, DePeter KC, Feldman HA, Grace E, Emans SJ. Prevalence of vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med 2004;158:531-7.
- Hypponen E, Power C. Hypovitaminosis D in British adults at age 45 y: nationwide cohort study of dietary and lifestyle predictors. Am J Clin Nutr 2007;85:860-8.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, Dawson-Hughes B, Garland CF, Heaney RP, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007;85:649-50.
- Zittermann A. Vitamin D in preventive medicine: are we ignoring the evidence? Br J Nutr 2003;89:552-72.
- Bodnar LM, Simhan HN, Powers RW, Frank MP, Cooperstein E, Roberts JM. High prevalence of vitamin D insufficiency in black and white pregnant women residing in the northern United States and their neonates. J Nutr 2007;137:447-52.
- Gonzalez G, Alvarado JN, Rojas A, Navarrete C, Velasquez CG, Arteaga E. High prevalence of vitamin D deficiency in Chilean healthy postmenopausal women with normal sun exposure: additional evidence for a worldwide concern. Menopause 2007;14:455-61.
- Siddiqui AM, Kamfar HZ. Prevalence of vitamin D deficiency rickets in adolescent school girls in Western region, Saudi Arabia. Saudi Med J 2007;28:441-4.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Ster Biochem Mol Biol 2007;103:204-5.
- Omdahl JL, Morris HA, May BK. Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr 2002;22:139-66.
- Hollis BW, Wagner CL, Drezner MK, Binkley NC. Circulating vitamin D3 and 25-hydroxyvitamin D in humans: An important tool to define adequate nutritional vitamin D status. J Steroid Biochem Mol Biol 2007;103:631-4.
- Kemmis CM, Salvador SM, Smith KM, Welsh J. Human mammary epithelial cells express CYP27B1 and are growth inhibited by 25-hydroxyvitamin D-3, the major circulating form of vitamin D-3. J Nutr 2006;136:887-92.
- Townsend K, Banwell CM, Guy M, Colston KW, Mansi JL, Stewart PM, et al. Autocrine metabolism of vitamin D in normal and malignant breast tissue. Clin Cancer Res 2005;11:3579-86.
- Townsend K, Evans KN, Campbell MJ, Colston KW, Adams JS, Hewison M. Biological actions of extra-renal 25-hydroxyvitamin D-1alpha-hydroxylase and implications for chemoprevention and treatment. J Steroid Biochem Mol Biol 2005;97:103-9.
- Zinser GM, Welsh JE. Accelerated mammary gland development during pregnancy and delayed post-lactational involution in vitamin D3 receptor null mice. Mol Endocrinol 2004;18:2208-23.
- Willnow TE, Nykjaer A. Pathways for kidney-specific uptake of the steroid hormone 25-hydroxyvitamin D3. Curr Opin Lipidol 2002;13:255-60.
- Norman AW. Minireview: vitamin D receptor: new assignments for an already busy receptor. Endocrinology 2006;147:5542-8.
- Frampton RJ, Suva LJ, Eisman JA, Findlay DM, Moore GE, Moseley JM, et al. Presence of 1,25-dihydroxyvitamin D3 receptors in established human cancer cell lines in culture. Cancer Res 1982;42:1116-9.
- Krishnan AV, Shinghal R, Raghavachari N, Brooks JD, Peehl DM, Feldman D. Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA micro-arrays. Prostate 2004;59:243-51.
- Lin R, Nagai Y, Sladek R, Bastien Y, Ho J, Petrecca K, et al. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol Endocrinol 2002;16:1243-56.
- Swami S, Raghavachari N, Muller UR, Bao YP, Feldman D. Vitamin D growth inhibition of breast cancer cells: gene expression patterns assessed by cDNA microarray. Breast Cancer Res Treat 2003;80:49-62.
- Towsend K, Trevino V, Falciani F, Stewart PM, Hewison M, Campbell MJ. Identification of VDR-responsive gene signatures in breast cancer cells. Oncology 2006;71:111-23.
- Moore DD, Kato S, Xie W, Mangelsdorf DJ, Schmidt DR, Xiao R, et al. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol Rev 2006;58:742-59.
- Carlberg C. Current understanding of the function of the nuclear vitamin D receptor in response to its natural and synthetic ligands. Recent Results Cancer Res 2003;164:29-42.
- Danielsson C, Mathiasen IS, James SY, Nayeri S, Bretting C, Hansen CM, et al. Sensitive induction of apoptosis in breast cancer cells by a novel 1,25-dihydroxyvitamin D3 analogue shows relation to promoter selectivity. J Cell Biochem 1997;66:552-62.
- Ellison TI, Eckert RL, MacDonald PN. Evidence for 1,25-dihydroxyvitamin D3-independent transactivation by the vitamin D receptor: uncoupling the receptor and ligand in keratinocytes. J Biol Chem 2007;282:10,953-62.
- Inoue T, Kamiyama J, Sakai T. Sp1 and NF-Y synergistically mediate the effect of vitamin D(3) in the p27(Kip1) gene promoter that lacks vitamin D response elements. J Biol Chem 1999;274:32309-17.
- Ellison TI, Dowd DR, MacDonald PN. Calmodulin-dependent kinase IV stimulates vitamin D receptor-mediated transcription. Mol Endocrinol 2005;19:2309-19.
- Jurutka PW, MacDonald PN, Nakajima S, Hsieh JC, Thompson PD, Whitfield GK, et al. Isolation of baculovirus-expressed human vitamin D receptor: DNA responsive element interactions and phosphorylation of the purified receptor. J Cell Biochem 2002;85:435-57.
- Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz J, et al. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1α,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci USA 2004;101:12876-81.
- Zanello LP, Norman AW. Rapid modulation of osteoblast ion channel responses by 1(alpha),25(OH)2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci USA 2004;101:12876-81.
- Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1{alpha},25(OH)2-vitamin D3 in vivo and in vitro. Mol Endocrinol 2004;18:2660-71.
- Bettoun DJ, Buck DW, Lu J, Khalifa B, Chin WW, Nagpal S. A vitamin D receptor-Ser/Thr phosphatase-p70 S6 kinase complex and modulation of its enzymatic activities by the ligand. J Biol Chem 2002;277:24847-50.
- Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell 2006;21:799-809.
- Wali RK, Kong J, Sitrin MD, Bissonnette M, Li YC. Vitamin D receptor is not required for the rapid actions of 1,25-dihydroxyvitamin D3 to increase intracellular calcium and activate protein kinase C in mouse osteoblasts. J Cell Biochem 2003;88:794-801.
- Khanal R, Nemere I. Membrane receptors for vitamin D metabolites. Crit Rev Eukaryot Gene Expr 2007;17:31-47.
- Bouillon R, Eelen G, Verlinden L, Mathieu C, Carmeliet G, Verstuyf A. Vitamin D and cancer. J Steroid Biochem Mol Biol 2006;102:156-62.
- Bouillon R, Verlinden L, Eelen G, De Clercq P, Vandewalle M, Mathieu C, et al. Mechanisms for the selective action of Vitamin D analogs. J Steroid Biochem Mol Biol 2005;97:21-30.
- Mork-Hansen C, Binderup L, Hamberg KJ, Carlberg C. Vitamin D and cancer: effects of 1,25(OH)2D3 and its analogs on growth control and tumorigenesis. Front Biosci 2001;6:D820-48.
- Munker R, Kobayashi T, Elstner E, Norman AW, Uskokovic M, Zhang W, et al. A new series of vitamin D analogs is highly active for clonal inhibition, differentiation, and induction of WAF1 in myeloid leukemia. Blood 1996;88:2201-9.
- Eisman JA, Sutherland RL, McMenemy ML, Fragonas JC, Musgrove EA, Pang GY. Effects of 1,25-dihydroxyvitamin D3 on cell-cycle kinetics of T 47D human breast cancer cells. J Cell Physiol 1989;138:611-16.
- Lazzaro G, Agadir A, Qing W, Poria M, Mehta RR, Moriarty RM, et al. Induction of differentiation by 1alpha-hydroxyvitamin D(5) in T47D human breast cancer cells and its interaction with vitamin D receptors. Eur J Cancer 2000;36:780-6.
- Narvaez CJ, Welsh J. Role of mitochondria and caspases in vitamin D-mediated apoptosis of MCF-7 breast cancer cells. J Biol Chem 2001;276:9101-7.
- Pendas-Franco N, Gonzalez-Sancho JM, Suarez Y, Aguilera O, Steinmeyer A, Gamallo C, et al. Vitamin D regulates the phenotype of human breast cancer cells. Differentiation 2007;75:193-207.
- Simboli-Campbell M, Narvaez CJ, vanWeelden K, Tenniswood M, Welsh J. Comparative effects of 1,25(OH)2D3 and EB1089 on cell cycle kinetics and apoptosis in MCF-7 breast cancer cells. Breast Cancer Res Treat 1997;42:31-41.
- Hager G, Formanek M, Gedlicka C, Thurnher D, Knerer B, Kornfehl J. 1,25(OH)2 vitamin D3 induces elevated expression of the cell cycle-regulating genes P21 and P27 in squamous carcinoma cell lines of the head and neck. Acta Otolaryngol 2001;121:103-9.
- Wu G, Fan RS, Li W, Ko TC, Brattain MG. Modulation of cell cycle control by vitamin D3 and its analogue, EB1089, in human breast cancer cells. Oncogene 1997;15:1555-63.
- Cordero JB, Cozzolino M, Lu Y, Vidal M, Slatopolsky E, Stahl PD, et al. 1,25-Dihydroxyvitamin D down-regulates cell membrane growth- and nuclear growth-promoting signals by the epidermal growth factor receptor. J Biol Chem 2002;277:38965-71.
- Kawata H, Kamiakito T, Takayashiki N, Tanaka A. Vitamin D3 suppresses the androgen-stimulated growth of mouse mammary carcinoma SC-3 cells by transcriptional repression of fibroblast growth factor 8. J Cell Physiol 2006;207:793-9.
- Wu Y, Craig TA, Lutz WH, Kumar R. Identification of 1 alpha,25-dihydroxyvitamin D3 response elements in the human transforming growth factor beta 2 gene. Biochemistry 1999;38:2654-60.
- Xie SP, Pirianov G, Colston KW. Vitamin D analogues suppress IGF-I signalling and promote apoptosis in breast cancer cells. Eur J Cancer 1999;35:1717-23.
- Mathiasen IS, Lademann U, Jaattela M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res 1999;59:4848-56.
- Chaudhry M, Sundaram S, Gennings C, Carter H, Gewirtz DA. The vitamin D3 analog, ILX-23-7553, enhances the response to adriamycin and irradiation in MCF-7 breast tumor cells. Cancer Chemother Pharmacol 2001;47:429-36.
- Posner GH, Crawford KR, Peleg S, Welsh JE, Romu S, Gewirtz DA, et al. A non-calcemic sulfone version of the vitamin D(3) analogue seocalcitol (EB 1089): chemical synthesis, biological evaluation and potency enhancement of the anticancer drug adriamycin. Bioorg Med Chem 2001;9:2365-71.
- Sundaram S, Gewirtz DA. The vitamin D3 analog EB 1089 enhances the response of human breast tumor cells to radiation. Radiat Res 1999;152:479-86.
- Sundaram S, Sea A, Feldman S, Strawbridge R, Hoopes PJ, Demidenko E, et al. The Combination of a Potent Vitamin D(3) Analog, EB 1089, with Ionizing Radiation Reduces Tumor Growth and Induces Apoptosis of MCF-7 Breast Tumor Xenografts in Nude Mice. Clin Cancer Res 2003;9:2350-6.
- Mathiasen IS, Sergeev IN, Bastholm L, Elling F, Norman AW, Jaattela M. Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J Biol Chem 2002;277:30738-45.
- Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 2005;12:1297-309.
- Simboli-Campbell M, Narvaez CJ, Tenniswood M, Welsh J. 1,25-Dihydroxyvitamin D3 induces morphological and biochemical markers of apoptosis in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol 1996;58:367-76.
- Valrance ME, Brunet AH, Acosta A, Welsh J. Dissociation of growth arrest and CYP24 induction by VDR ligands in mammary tumor cells. J Cell Biochem 2007. in press.
- Kommagani R, Caserta TM, Kadakia MP. Identification of vitamin D receptor as a target of p63. Oncogene 2006;25:3745-51.
- Maruyama R, Aoki F, Toyota M, Sasaki Y, Akashi H, Mita H, et al. Comparative genome analysis identifies the vitamin D receptor gene as a direct target of p53-mediated transcriptional activa-tion. Cancer Res 2006;66:4574-83.
- Saramaki A, Banwell CM, Campbell MJ, Carlberg C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res 2006;34:543-54.
- Zinser GM, McEleney K, Welsh J. Characterization of mammary tumor cell lines from wild type and vitamin D(3) receptor knockout mice. Mol Cell Endocrinol 2003;200:67-80.
- Flanagan L, Packman K, Juba B, O’Neill S, Tenniswood M, Welsh J. Efficacy of Vitamin D compounds to modulate estrogen receptor negative breast cancer growth and invasion. J Steroid Biochem Mol Biol 2003;84:181-92.
- James SY, Mercer E, Brady M, Binderup L, Colston KW. EB1089, a synthetic analogue of vitamin D, induces apoptosis in breast cancer cells in vivo and in vitro. Br J Pharmacol 1998;125:953-62.
- VanWeelden K, Flanagan L, Binderup L, Tenniswood M, Welsh J. Apoptotic regression of MCF-7 xenografts in nude mice treated with the vitamin D3 analog, EB1089. Endocrinology 1998;139:2102-10.
- Eisman JA, Macintyre I, Martin TJ, Frampton RJ, King RJ. Normal and malignant breast tissue is a target organ for 1,25-(OH)2 vitamin D3. Clin Endocrinol 1980;13:267-72.
- Zinser G, Packman K, Welsh J. Vitamin D(3) receptor ablation alters mammary gland morphogenesis . Development 2002;129:3067-76.
- Zinser GM, Welsh J. Accelerated mammary gland development during pregnancy and delayed postlactational involution in vitamin D3 receptor null mice. Mol Endocrinol 2004;18:2208-23.
- Rowling MJ, Kemmis CM, Taffany DA, Welsh J. Megalin-mediated endocytosis of vitamin D binding protein correlates with 25-hydroxycholecalciferol actions in human mammary cells. J Nutr 2006;136:2754-9.
- Lipkin M, Newmark HL. Vitamin D, calcium and prevention of breast cancer: a review. J Am Coll Nutr 1999;18:392S-7S.
- Mehta RG, Moriarty RM, Mehta RR, Penmasta R, Lazzaro G, Constantinou A, et al. Prevention of preneoplastic mammary lesion development by a novel vitamin D analogue, 1alpha-hydroxyvitamin D5. J Natl Cancer Inst 1997;89:212-18.
- Bernardi RJ, Johnson CS, Modzelewski RA, Trump DL. Antiproliferative effects of 1alpha,25-dihydroxyvitamin D(3) and vitamin D analogs on tumor-derived endothelial cells. Endocrinology 2002;143:2508-14.
- El Abdaimi K, Dion N, Papavasiliou V, Cardinal PE, Binderup L, Goltzman D, et al. The vitamin D analogue EB1089 prevents skeletal metastasis and prolongs survival time in nude mice transplanted with human breast cancer cells. Cancer Res 2000;60:4412-8.
- Zinser GM, Suckow M, Welsh J. Vitamin D receptor (VDR) ablation alters carcinogen-induced tumorigenesis in mammary gland, epidermis and lymphoid tissues. J Steroid Biochem Mol Biol 2005;97:153-64.
- Zinser GM, Welsh J. Vitamin D receptor status alters mammary gland morphology and tumorigenesis in MMTV-neu mice. Carcinogenesis 2004;25:2361-72.
- Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002;23:2103-9.
- Agadir A, Lazzaro G, Zheng Y, Zhang XK, Mehta R. Resistance of HBL100 human breast epithelial cells to vitamin D action. Carcinogenesis 1999;20:577-82.
- Solomon C, Kremer R, White JH, Rhim JS. Vitamin D resistance in RAS-transformed keratinocytes: mechanism and reversal strategies. Radiat Res 2001;155:156-62.
- Hansen CM, Rohde L, Madsen MW, Hansen D, Colston KW, Pirianov G, et al. MCF-7/VD(R): a new vitamin D resistant cell line. J Cell Biochem 2001;82:422-36.
- Narvaez CJ, Vanweelden K, Byrne I, Welsh J. Characterization of a vitamin D3-resistant MCF-7 cell line. Endocrinology 1996;137:400-9.
- Byrne B, Welsh J. Identification of novel mediators of Vitamin D signaling and 1,25(OH)2D3 resistance in mammary cells. J Steroid Biochem Mol Biol 2007;103:703-7.
- Narvaez CJ, Byrne BM, Romu S, Valrance M, Welsh J. Induction of apoptosis by 1,25-dihydroxyvitamin D(3) in MCF-7 Vitamin D(3)-resistant variant can be sensitized by TPA. J Steroid Biochem Mol Biol 2003;84:199-209.
- Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, et al. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet 2000;25:144-6.
- Abedin SA, Banwell CM, Colston KW, Carlberg C, Campbell MJ. Epigenetic corruption of VDR signalling in malignancy. Anticancer Res 2006;26:2557-66.
- Banwell CM, MacCartney DP, Guy M, Miles AE, Uskokovic MR, Mansi J, et al. Altered nuclear receptor corepressor expression attenuates vitamin D receptor signaling in breast cancer cells. Clin Cancer Res 2006;12:2004-13.
- Knekt P, Jarvinen R, Seppanen R, Pukkala E, Aromaa A. Intake of dairy products and the risk of breast cancer. Br J Cancer 1996;73:687-91.
- John EM, Schwartz GG, Dreon DM, Koo J. Vitamin D and breast cancer risk: the NHANES I Epidemiologic follow-up study, 1971-1975 to 1992. National Health and Nutrition Examination Survey. Cancer Epidemiol Biomarkers Prev 1999;8:399-406.
- Lin J, Manson JE, Lee IM, Cook NR, Buring JE, Zhang SM. Intakes of calcium and vitamin d and breast cancer risk in women. Arch Intern Med 2007;167:1050-9.
- Berube S, Diorio C, Verhoek-Oftedahl W, Brisson J. Vitamin D, calcium and mammographic breast densities. Cancer Epidemiol Biomarkers Prev 2004;13:1466-72.
- Brisson J, Berube S, Diorio C, Sinotte M, Pollak M, Masse B. Synchronized seasonal variations of mammographic breast density and plasma 25-hydroxyvitamin d. Cancer Epidemiol Biomarkers Prev 2007;16:929-33.
- Giovannucci E. The epidemiology of vitamin D and cancer incidence and mortality: a review (United States). Cancer Causes Control 2005;16:83-95.
- Grant WB, Garland CF. The association of solar ultraviolet B (UVB) with reducing risk of cancer: multifactorial ecologic analysis of geographic variation in age-adjusted cancer mortality rates. Anticancer Res 2006;26:2687-99.
- Garland CF, Gorham ED, Mohr SB, Grant WB, Giovannucci EL, Lipkin M, et al. Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol 2007;103:708-11.
- Glerup H, Mikkelsen K, Poulsen L, Hass E, Overbeck S, Thomsen J, et al. Commonly recommended daily intake of vitamin D is not sufficient if sunlight exposure is limited. J Intern Med 2000;247:260-8.
- Hart GR, Furniss JL, Laurie D, Durham SK. Measurement of vitamin D status: background, clinical use, and methodologies. Clin Lab 2006;52:335-43.