
Predicting the physiological relevance of in vitro cancer preventive activities of phytochemicals1
Introduction
In recent years there has been increasing interest in the use of biologically active phytochemicals in cancer prevention. In particular, many in vitro studies using a wide range of natural products have demonstrated a preferential induction of cell cycle arrest or apoptosis in tumor cell lines compared to lines derived from non-tumor tissue. On further investigation, phytochemicals have been found to modulate the expression or activity of a large number of cellular proteins which are key for cell survival and the transformed phenotype. However, there is also much concern that many of these effects[1–3] are irrelevant in vivo, since the concentrations used are often orders of magnitude greater than appear to be achievable in the human body.
In this review, we have attempted to address this concern for a few of the most studied diet-derived compounds. For each agent, we have assimilated reported in vivo concentrations, based where possible on human data. We have then surveyed the in vitro biological effects at these or lower doses. For the indoles only, we have included data on phase I drug metabolizing activity relating to altered estrogen metabolism. Encouragingly, a significant amount of published data validates some of these changes in vivo.
For a number of reasons, some guesswork was involved in deciding on the most relevant in vitro doses for consideration. First, adding a compound directly to a cell culture may deliver a much higher local dose than occurs following ingestion in the body. Second, some if not all of the compounds may undergo metabolism in vivo to other more or less active derivatives and such metabolism may not be possible in culture. Third, higher in vivo doses than those so far reported may be achievable by administration of a pure compound rather than a dietary source, or an optimized formulation of a pure compound. Fourth, some target tissues may receive a higher (or more prolonged) dose than the reported peak levels in plasma. Fifth, some target tissues, such as skin, oral cavity, gastrointestinal tract, and bladder, may receive higher doses because they are not dependent on circulating levels. The colon, for example, can be exposed to significant amounts of (unabsorbed) excreted material and the lining of the bladder may be exposed for substantial periods of time to any compounds concentrated in urine. Finally, where diet is concerned, any one compound may be poorly bioavailable, but with dozens, even hundreds, of active molecules being ingested together, the cumulative dose of similar acting compounds may be significantly higher.
It should not be forgotten that some natural products have exhibited toxicity in vivo when given in high doses, so there is also an argument for the use of higher concentrations in vitro to indicate the full range of activity of these molecules.
Indole-3-carbinol and diindolylmethane
Indole-3-carbinol (I3C) is derived from glucobrassicin, found in cruciferous vegetables. Diindolylmethane (DIM) is an acid condensation product formed from 2 molecules of I3C. In vivo, this is thought to occur in the acid conditions of the stomach. Data for mammals only are considered here.
Bioavailability of I3C and DIM in humans Information on the bioavailability and tissue distribution of I3C or DIM in humans is very limited[4–6]. However, there are a number of studies in which oral administration of I3C resulted in a biochemically or clinically measurable outcome, indicating that the absorption of I3C and/or its acid-condensation products does occur. Administered doses of I3C have ranged from 200 to 500 mg/d (~ 6–7 mg/kg), typically for periods of 1–6 months, although treatments up to 82 months have also been reported[7–14]. A dose-dependent effect (placebo, 200 and 400 mg/d for 3 months) was observed in the treatment of cervical intraepithelial neoplasia[7], and 200 or 400 mg/d were similarly effective against vulval intraepithelial neoplasia[14]. In dose-escalation studies for breast cancer prevention, 300 mg/d (minimum) increased the urinary estrogen metabolite ratio of 2-hydroxyestrone to 16 alpha-hydroxyestrone; 800 mg/d did not provide additional benefits over 400 mg/d in adult women[15,16]. Elevated cytochrome P450 activity was responsible for an increase in 2-hydroxylation of estrogen, increasing the ratio of 2-OH:16-OH estrone[16], which is regarded as favorable for the prevention of breast cancer and human papilloma virus (HPV)-related neoplasias[7–14].
Two studies detected DIM in plasma (2.5 µmol/L maximum at 2 h, gradually decreasing by 12 h) or in urine following oral administration of I3C (Table 1). I3C was not detected in plasma or serum following oral doses of 400–1200 mg[4,5]. DIM was also detected in the urine of a patient receiving DIM[4]. In a pilot study using a formulation of enhanced absorption DIM (BioResponse DIM, 108 mg/d for 30 d), increased 2-hydroxylation of estrogen was also reported[6].

Full table
Bioavailability of I3C and DIM in animals Radio-labeled I3C was used to follow distribution and tissue content in several studies, although this method did not differentiate between I3C and related products. In rats receiving 50 mg 14C-I3C by gavage, I3C equivalents peaked at 28 µmol/L in the blood and 121 µmol/L in the liver after 30 min. The labeled product was detectable in the 100 µmol/L range from 10 min to 2 h following dosing[17]. High maximal concentrations of the I3C equivalent, but not I3C itself, were detected in the liver (1154 µmol/L), kidney, lung (436 µmol/L), blood (320 µmol/L), and tongue of the rats given 3H-I3C in the diet for 1 week (0.88+/–0.074 mmol/kg/d)[18]. Once a steady state had been reached, excretion in feces and urine accounted for 75% dose/d, the majority of this being present in the feces by 110 h, indicating that either the dose was not absorbed or that a major excretory route was via bile. When 14C-I3C was given to pregnant mice, it was detected in the fetal liver, stomach, kidney, intestine, and lung (100–300 µmol/L) after 8 h of maternal exposure[19].
Anderton et al detected I3C and DIM in tissues following the dosing of mice with 250 mg/kg I3C (Table 2) using an HPLC method allowing the simultaneous identification and quantification of I3C and its derivatives[20]. The maximum level of 28 µmol/L I3C was observed at 15 min, falling below the level of detection by 1 h after dosing. I3C was detected in the liver (170 µmol/L)>kidney (116 µmol/L)>lung and heart>plasma>brain. The levels of DIM peaked at around 2 h in the liver (16 µmol/L)>lung and kidney>heart>brain>plasma (4 μmol/L), and by 24 h, were still detectable at approximately 0.5 µg/g in the brain and liver. The presence of linear trimer, 1-(3-hydroxymethyl)-indolyl-3-indolylmethane and indolo(3,2b)carbazole, together with oxidative metabolites of I3C, was also documented. In a further study, Anderton et al compared concentrations of DIM in tissues of mice dosed with either pure DIM (250 mg/kg; Table 2) or an equivalent dose of the enhanced absorption BioResponse DIM[21]. The tissue distribution of DIM was similar to that reported previously following the administration of I3C with maximal concentrations around 160 µmol/L in the liver[20]. The BioRes-ponse DIM resulted in levels approximately 50% higher than those obtained with unformulated DIM.

Full table
Thus, following the oral administration of I3C, both I3C and DIM were detectable at µmol/L concentrations in the blood and multiple organs. I3C was rapidly absorbed and cleared from the blood and tissues within 1 h, while DIM peaked slightly later and was more persistent. The observation of I3C in the blood and tissues at these very early time points belies previous assumptions that I3C is not absorbed, but undergoes complete acid condensation in the stomach. Several studies have revealed distinct responses to I3C and DIM in animal models[22–26]. Therefore, the in vivo activity of dietary I3C cannot be attributed completely to the production of DIM, although response due partially to DIM conversion is probable.
Physiologically relevant concentrations of I3C and DIM As no data are available for achievable levels of I3C in humans, we extrapolated from animal studies[20,21]. The maximum plasma and tissue concentrations attained in mice for I3C were 28 and 170 µmol/L (15 min) and for DIM, 4 and 16 µmol/L (2 h), respectively (Table 2). By allometric scaling, the I3C dose given to mice would equate to a 20 mg/kg dose in humans (1200 mg/d), which yielded the maximal serum concentration 2.5 µmol/L DIM (2 h), with no I3C detectable after 1 h[5]. Therefore, maximal detectable DIM concentrations following I3C administration are similar in mice and humans, and the discrepancy in I3C detection is likely to be caused by the sensitivity of methods and selection of time points.
The maximum levels of DIM achieved in animals following a dose of a pure compound range from 24–200 µmol/L (Table 2), with BioResponse DIM resulting in 50% higher bioavailability. The dose of BioResponse DIM used in humans was 108 mg (~1.3–1.9 mg/kg)[6], which might be expected to give levels in the range 3–30 µmol/L.
For the purposes of this review, the effects of the physiological concentrations up to 150 µmol/L I3C and 50 µmol/L DIM in vitro have been considered.
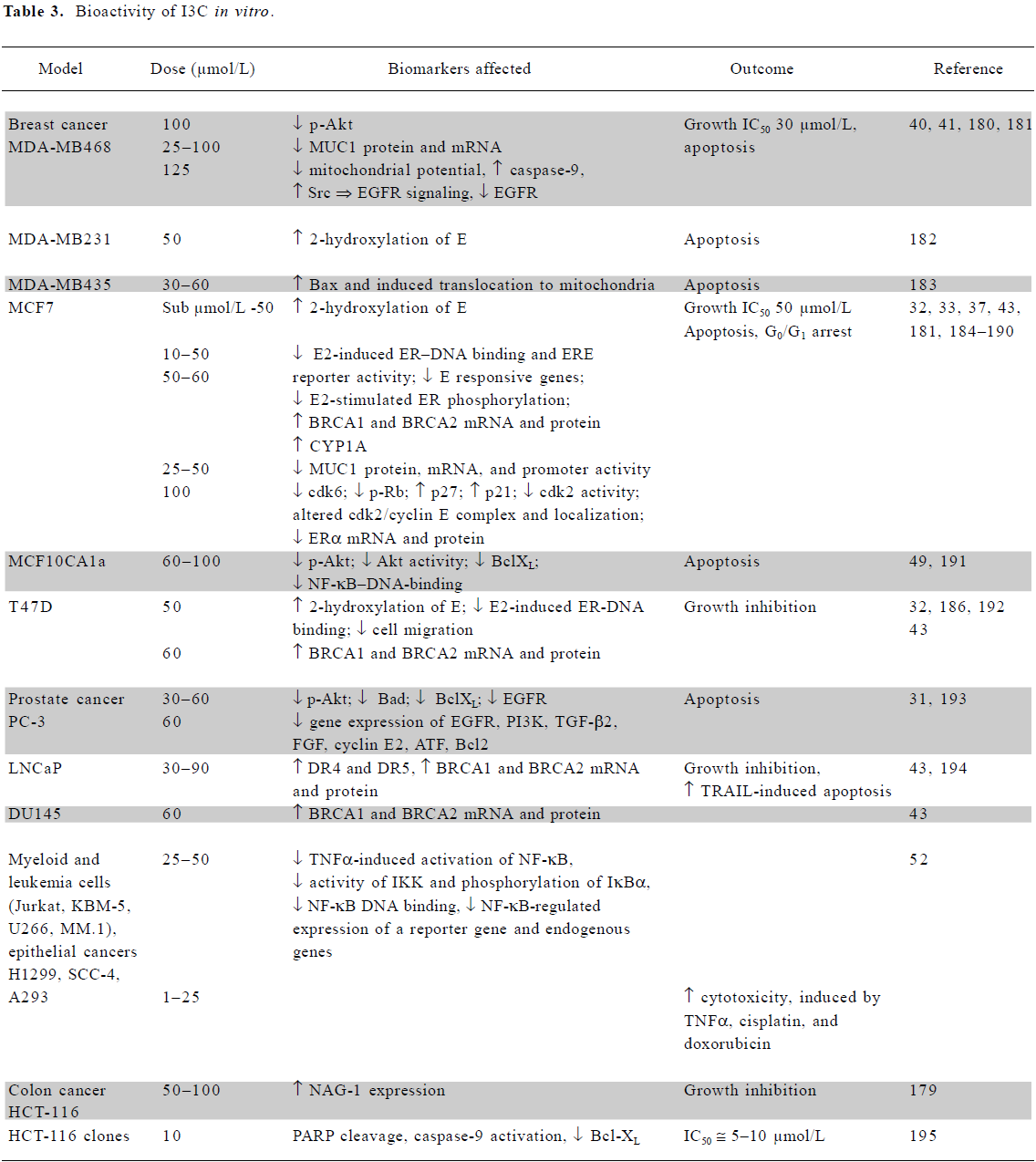
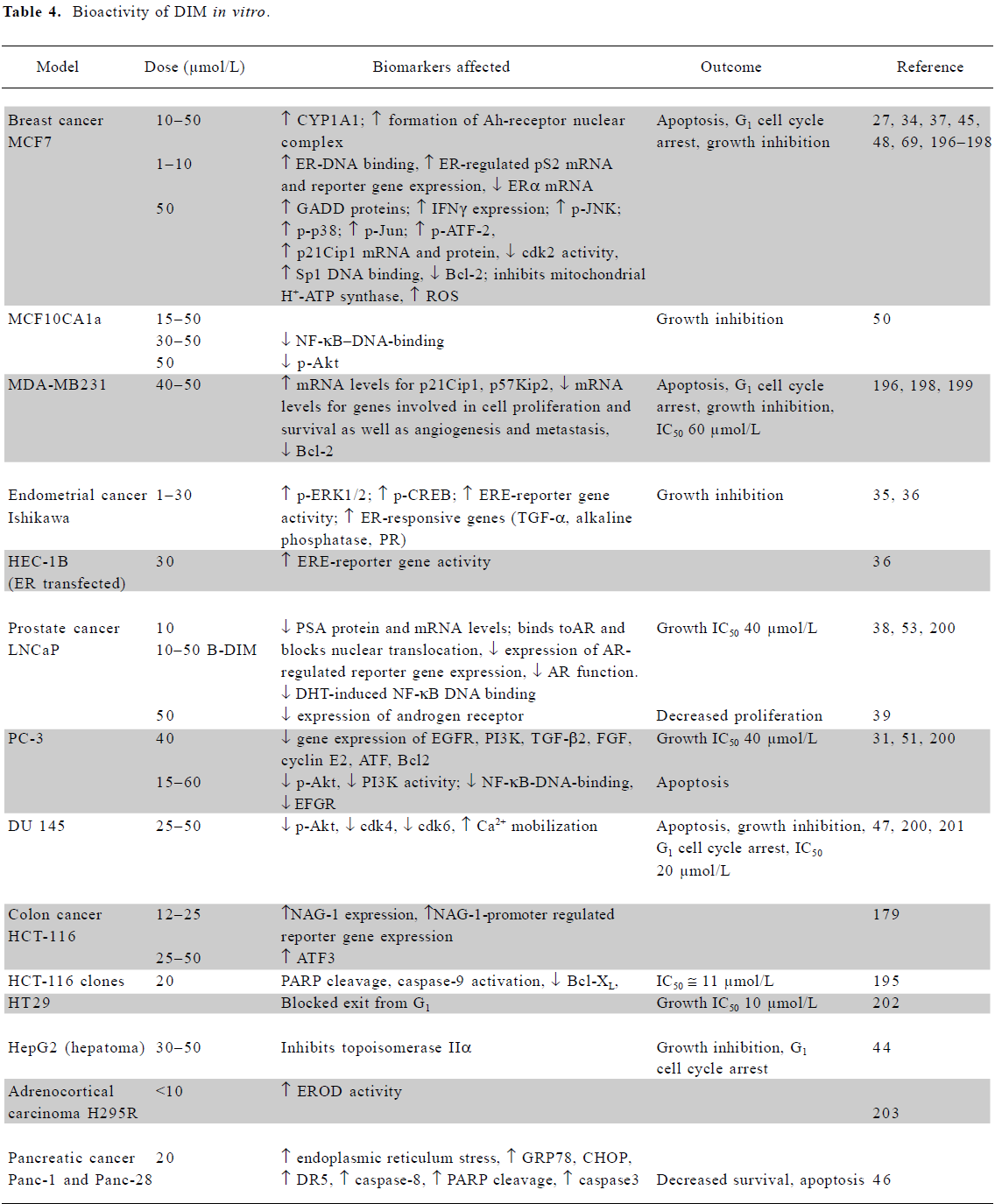
In vitro mechanistic studies using low doses of I3C or DIM Several mechanisms are responsible for the chemo-preventive activities of I3C and DIM, as summarized in Tables 3 and 4. Both agents induce activity of phase I and II enzymes involved in the biotransformation and elimination of carcinogens and steroid hormones. While detailed molecular interactions involved have not been completely elucidated, DIM interacts with the aryl hydrocarbon receptor (AhR), resulting in its nuclear translocation and induction of the genes encoding phase1 and II enzymes[27]. Several lines of evidence suggest that DIM exerts agonist and/or modulator activity on the AhR[28,29]. I3C can activate the NF-E2-related factor-2 (Nrf2) transcription factor which interacts with the antioxidant response element in the promoter of many cytoprotective enzymes, as described later. The induction of cytochrome P450 (CYP450) by physiological concentrations of I3C and DIM was observed in cancer cells[30] and confirmed by an analysis of mRNA expression profiles[31]. Increased CYP450 activity led to increased estrogen metabolism and the degradation of estradiol (E2)[27], which is required for the growth of estrogen receptor-alpha (ERα)-positive cancer cells. I3C (50 µmol/L) inhibited the estradiol-stimulated growth of estrogen-responsive MCF7, T47D and ZR75.1 breast and cervical cells. It inhibited receptor phosphorylation and DNA binding as well as estrogen-dependent reporter gene activity in breast tumor cells and cervical cancer cell lines[32,33]. DIM (10 µmol/L) also inhibited the estradiol-stimulated growth of MCF7, but in the absence of 17β-estradiol it appeared to stimulate growth[34].
Full table
Full table
DIM also exerted other estrogenic effects in these and endometrial cells[34–36]. Conversely, earlier data indicated an inhibitory effect of DIM on E2-regulated reporter activity and ER DNA binding[27]. Both I3C (75–100 µmol/L) and DIM (1–25 µmol/L) reduced the expression of ERα mRNA in MCF7 cells[33,37]. DIM also interfered with androgen receptor expression, DNA binding, and signaling[38,39].
Both compounds are growth inhibitory to a wide range of tumor cell lines, including ER-negative cancer cells. The increased loss of viability of several breast cancer cell lines when grown in a 3-D environment in the presence of I3C implied greater susceptibility in vivo than in a monolayer cell culture[40]. Recent studies have shown that I3C decreases proliferation and induces apoptosis by reducing the expression and signaling of the genes essential for tumor cell viability, such as ER and epidermal growth factor receptor (EGFR) in breast cells of luminal A and basal-like subtypes[33,41]. I3C (50 µmol/L) can also inhibit phosphoinositide-3-kinase (PI3K)[42], resulting in the inhibition of protein kinase B (Akt) phosphorylation and decreased survival in cancer cells dependent on this pathway. A mechanism dependent on breast cancer-related protein (BRCA)1/2 upregulation has also been proposed[43].
DIM, which is considerably more potent than I3C, also inhibits the growth of a range of cells (Table 4). Topoisomerase II-alpha and mitochondrial H+-ATP synthase were identified as direct targets of DIM. The inhibition of the latter enzyme results in increased mitochondrial reactive oxygen species (ROS) production and signaling via the p38 stress activation pathway[44,45]. Apoptosis via the activation of the endoplasmic reticulum stress pathway has also been reported[46,47].
Both agents have a significant effect on several other signaling pathways, such as those involving p38[45,48] and NF-κB signaling[49–53]. They modulate a variety of growth-, cell cycle-, and apoptotic-regulatory proteins at the mRNA or protein level, including EGFR, PI3K, transforming growth factor (TGF)-β2, fibroblast growth factor (FGF), cyclin E2, activating transcription factor (ATF), B cell lymphoma Bcl2, BclXL, Bad, and Bax.
In vitro effects of I3C or DIM observed in vivo Both I3C and DIM clearly inhibit tumor cell growth in vivo in a range of animal models[54]. The reduced incidence and multiplicity of mammary tumors was concurrent with increased phase I and II drug metabolizing enzymes in I3C-treated animals[55–57]. An analysis of transgenic Nrf2–/– mice indicated that the I3C-induced upregulation of phase II enzymes required the Nrf2 transcription factor[58,59]. The upregulation of CYP450 activity by DIM in vivo has been proposed to occur via a mechanism involving the AhR[30], as reported in MCF7 cells in vitro[27]. The induction of phase I and II enzymes has been reported in liver, small intestine, and lungs of rodents receiving I3C (in the diet or by gavage)[30,54,57,60–63]. DIM also induced P450 activity and flavin-containing monooxygenase 1 in the rat liver[30,60].
Apoptosis in response to I3C was observed in vivo in initiated mammary glands with activation of caspases-8, -9, and -3[25] and in cervical epithelium of transgenic mice (HPV16), developing cervical cancer in response to estrogen[56]. Few studies have investigated the effect of either agent on signal transduction intermediates in vivo, but in one study, dietary I3C (0.5%) caused a significant decrease in total tyrosine phosphorylation and ornithine decarboxylase activity in the rat liver[62]. Many of the signaling events modulated by I3C in vitro involve tyrosine phosphoryla-tion[41], but interestingly, changes in ornithine decarboxylase activity in breast and colon cells in vitro were only observed at relatively high concentrations (>100 µmol/L)[62,64]. The downregulation of NF-κB-regulated genes by I3C occurring in a variety of cancer cells in vitro, was also observed in mouse xenografts of MDA-MB231 cells[52,65].
Evidence for I3C or DIM acting synergistically/ antagonistically In rats, I3C (5 mg/kg) reversed vinblastine- or vincristine-induced P-glycoprotein levels[66]. This group also showed that a very high dose of I3C (10 mmol/L) decreased P-glycoprotein levels in vitro in the multidrug resistant cell line K562/R 10, sensitizing it to vinblastine, but had no growth-inhibitory effect on the parent K562 cell line[67]. I3C (333 or 500 mg/kg per day) also reversed the MDR phenotype of the B16/hMDR1 (drug-resistant MDR1-expressing murine melanoma) tumor in vivo, and in combination with vinblastine, actually reduced the tumor mass[68]. In the same study, an I3C acid-condensation mixture (12.5 µmol/L) sensitized the B16/hMDR1 cell line in vitro to vinblastine, while DIM (45 µmol/L) increased the drug content of cells by 50%[68]. Combined treatments using I3C (50–125 µmol/L) in combination with Src or/and EGFR inhibitors reduced the viability of breast cancer cells MDA-MB468 and MCF7[41].
DIM (25 µmol/L) plus genistein (5 µmol/L) synergistically induced growth arrest and DNA damage-inducible (GADD)34 protein levels and apoptosis, and at higher concentrations induced estrogen receptor response element (ERE)-driven reporter gene activity[69]. I3C (50 or 100 µmol/L) has been shown to cooperate with tamoxifen (1 µmol/L) in vitro to increase cell growth inhibition and G0/G1 cell cycle arrest of MCF7 cells[70]; in vivo, it reduces tumor mass and increases latency of mammary cancers[71]. An I3C acid condensation mixture also enhanced the efficacy of vinblastine in mouse melanoma cells, while I3C itself had no effect[68].
In vivo, dietary I3C, together with crambene (1-cyano 2-hydroxy 3-butene), another glucosinolate from vegetables, showed a greater than additive induction of glutathione-S-transferase (GST) and quinone reductase activity[72].
Curcumin
Curcumin (diferuloylmethane) is a major constituent of the spice turmeric, derived from the roots of Curcuma longa. The major dietary source is curry, but it is also used as a food coloring and in some medicines.
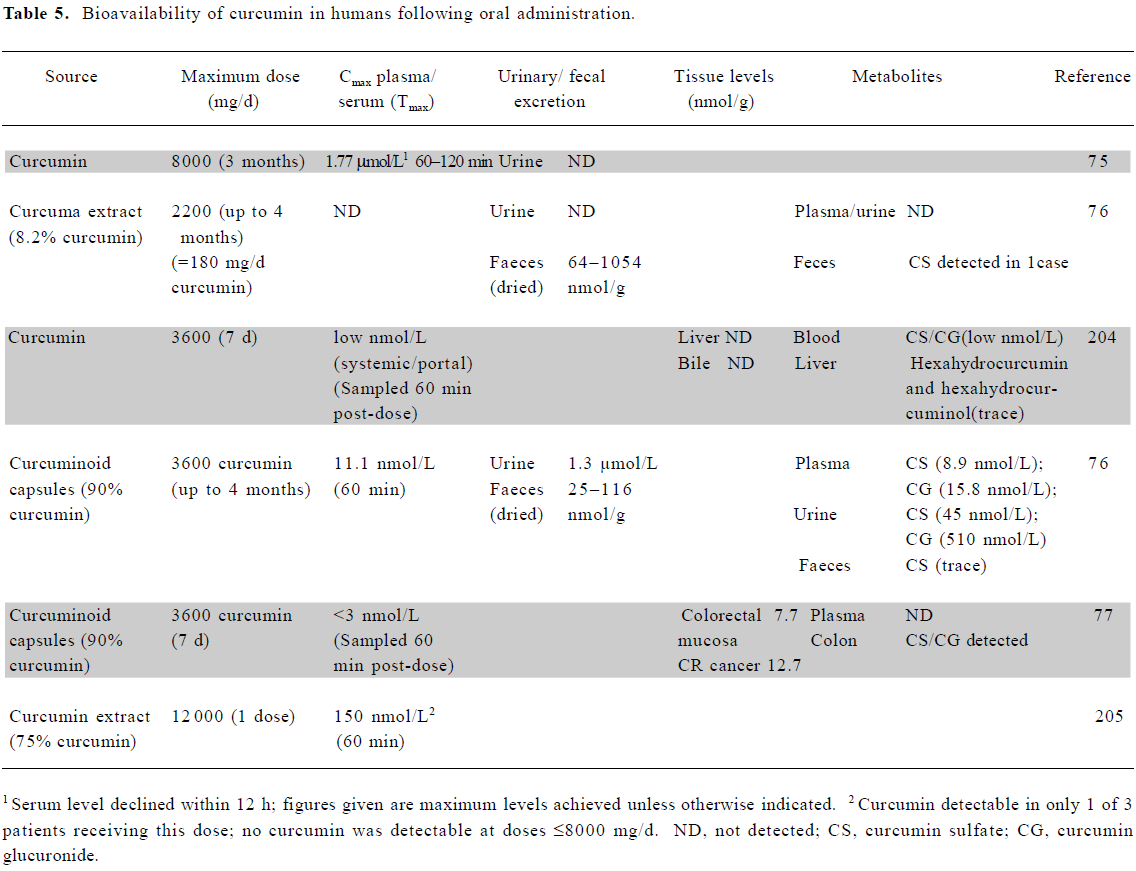
Bioavailability of curcumin in humans Curcumin exhibits poor gastrointestinal absorption, with much of an oral dose passing unchanged through the gastrointestinal tract, and a further proportion undergoing conjugation, without absorption, prior to fecal loss. Absorbed curcumin undergoes sequential reduction and conjugation (glucuronidation and/or sulfation) within the gastrointestinal tract and liver, with the resultant formation of metabolites and low systemic levels of the parent compound[73,74].
Studies in humans have demonstrated that the oral administration of curcumin furnishes very low systemic levels, mostly in the low nanomolar range (Table 5). An exception is the study by Cheng et al, which reported serum levels in the low micromolar range using the maximum tenable dose (8 g/d)[75]. Other groups have failed to replicate this finding, with Sharma et al, for example, administering up to 3.6 g/d of curcumin to patients for up to 4 months, yet only achieving levels in the 10 nmol/L range[76]. The discrepancy between these studies remains to be explained, but may have resulted from the use of different formulations of curcumin.
Full table
Due to its poor bioavailability, curcumin levels in tissues beyond the gastrointestinal tract are also in the low nanomolar range or below. Garcea et al were unable to detect curcumin in normal liver or colorectal liver metastases in patients who had received 3.6 g/d for 1 week. In the only human study to examine colorectal tissue to date, this oral dose resulted in levels in the 10 µmol/L range[77].
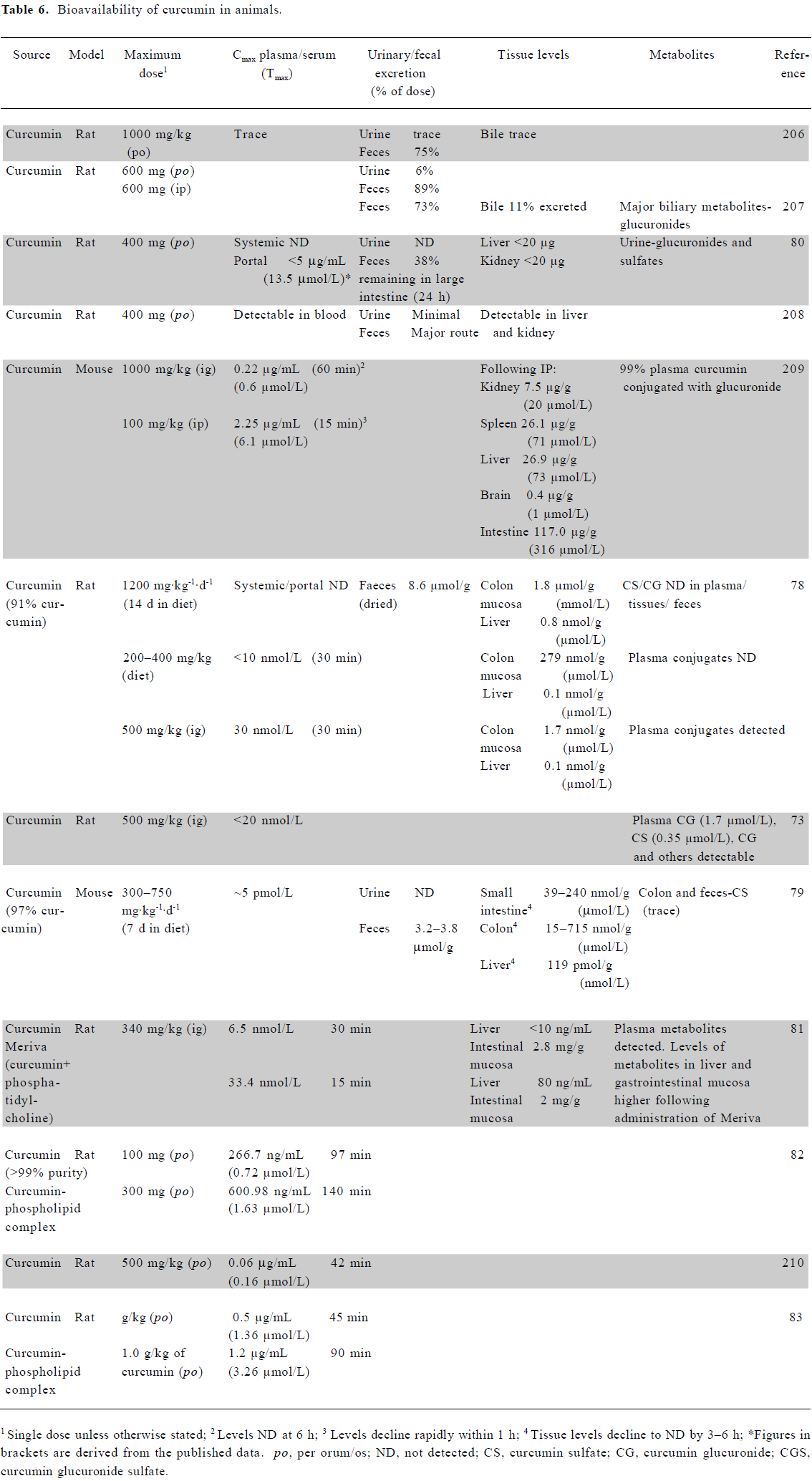
Bioavailability of curcumin in animals A number of groups have examined the bioavailability of curcumin in animals, following oral, intragastric (ig) or intraperitoneal (ip) dosing (Table 6). Studies suggest that oral dosing may give rise to significant levels of curcumin within the gastrointestinal tract. In a rat model, approximately 1.8 μmol curcumin/g of tissue was demonstrated in colonic mucosa following the dietary administration of 1200 mg/kg daily[78]. Perkins et al reported 750 mg/kg of curcumin/d to result in ~100 nmol/g in mouse small intestine mucosa and 500 nmol/g within colonic mucosa[79]. Following oral dosing of 400 mg per rat, liver and kidney levels were less than 20 μg per tissue[80]. Significant levels of curcumin may also be achieved locally when administered topically to the skin or within the oral cavity, but the exact dose achieved in these scenarios remains to be confirmed.
Full table
It is neither practicable nor desirable to increase the oral dose of curcumin above that already investigated. Recent animal studies, however, have demonstrated that the reformulation of curcumin may enable further improvements in bioavailability. It has previously been shown that the formulation of drugs with phosphatidylcholine increases their plasma bioavailability. Such a formulation led to significantly higher levels of curcumin within plasma and the liver compared with the parent compound, although lower levels within the intestinal mucosa[81]. Several other animal studies have also found curcumin bioavailability to be significantly increased by its administration as a phospholipid complex[82,83]. These increases in bioavailability now require confirmation in human studies. Although the bioavailability data are lacking, in vitro and animal studies have also shown promising anticancer potential for a liposomal preparation of curcumin[84,85]. In addition, nanoparticle-encapsulated curcumin may provide an alternative means to increase the bioavailability of this agent[86].
Physiologically relevant concentrations of curcumin The bioavailability data suggest that in vitro studies with curcumin in the 10 µmol/L range or below might have human physiological relevance, but that its role as a chemopreventive agent may lie primarily within the gastrointestinal tract.
In vitro mechanistic studies using low doses of curcumin The anticancer effects of curcumin have been demonstrated in multiple cell types, at concentrations between 5 and 50 µmol/L[87]. Selected studies demonstrating the anticancer activity of curcumin at or below the 10 µmol/L level achievable in the human colon in vivo are summarized in Table 7. Where available, data are presented from studies using colorectal cell lines; results from other cell types using a maximal dose of 10 µmol/L are also included. In addition to these studies, curcumin also inhibited the proliferation of squamous carcinoma SCC-25 cells[88] and the proliferation and invasion of HBL100 breast cells[89].
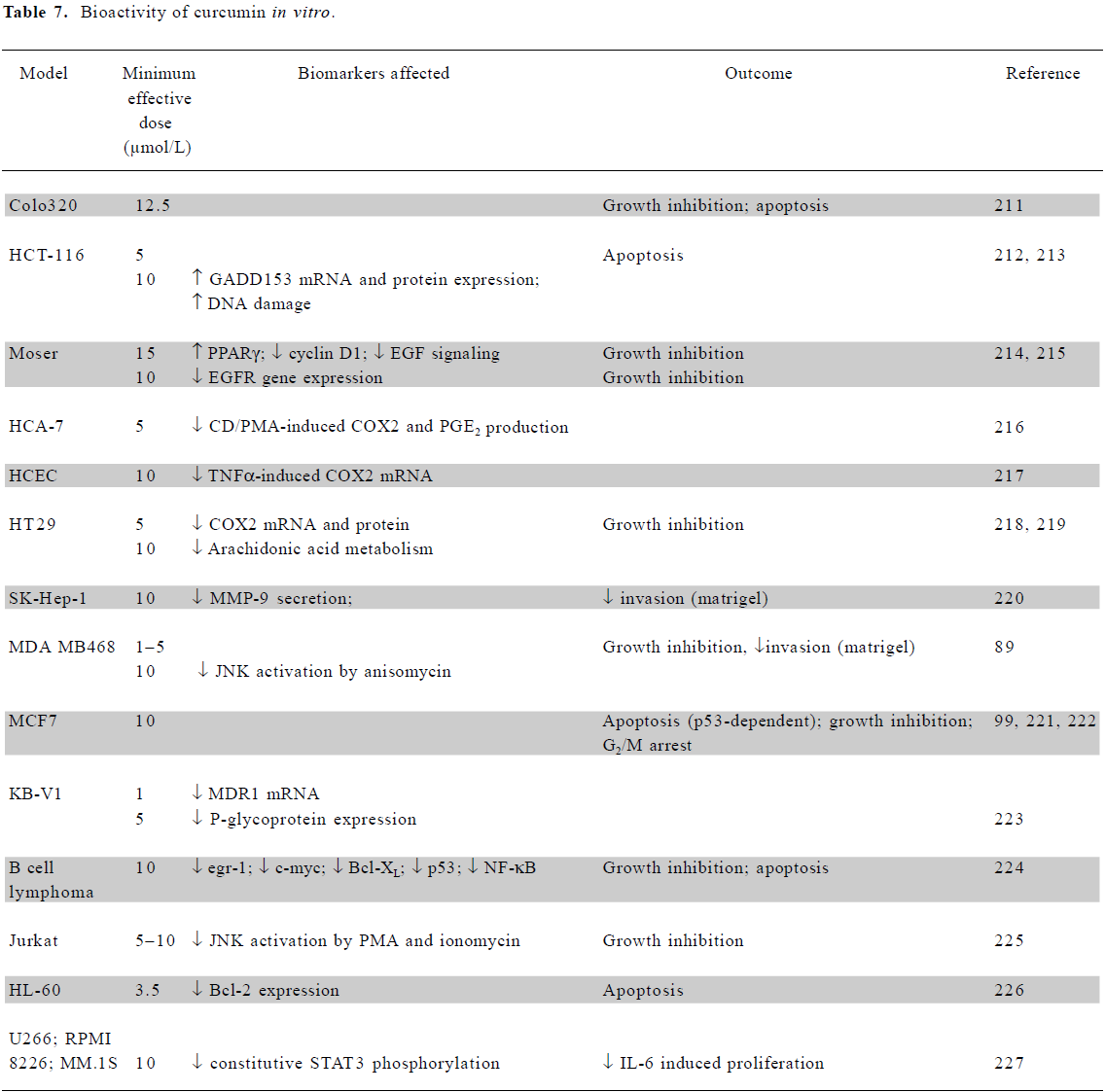
Full table
In vitro effects of curcumin observed in vivo In a rat model, dietary curcumin significantly increased the apoptotic index in azoxymethane-induced colonic tumors[90]. Rao et al demonstrated the effect of a curcumin-containing diet on azoxymethane-induced rat carcinogenesis[91]. Curcumin significantly reduced tumor volume, as well as colonic mucosa and tumor prostaglandin (PGE)2 expression by over 38%. Similarly, it enhanced 2-amino-1-methyl-6-phenylimidazol(4,5-b)pyridine-induced apoptosis in Min/+ mice and inhibited tumorigenesis in the proximal small intestine. Also in mice, Mahmoud et al found dietary curcumin to normalize enterocyte proliferation and restore the level of enterocyte apoptosis to that of wild-type animals[92]. In rats, a gavage administration of curcumin (200 or 600 mg/kg) inhibited diethylnitrosamine (DEN)-induced hepatic hyperplasia and inflammation. Specifically, the increased expression of p21ras and p53 in the liver was prevented. The decreased expression of proliferating cell nuclear antigen, cyclin E, and cdc2 was also observed, along with the inhibition of DEN-induced NF-κB activation[93].
While there are no in vitro studies for comparison, there is evidence from both animal and human studies showing that curcumin suppresses malondialdehyde-deoxyguanosine adduct (M1dG) adduct formation in DNA[77,78]. However, Garcea et al, while noting decreased M1dG adduct formation in the colorectum following curcumin treatment, found no alteration in cyclooxygenase 2 (COX2) protein levels[77].
Despite the low bioavailability of curcumin, there are examples in animal studies of its biological activity at sites distant from the locus of absorption, where levels are expected to be inefficacious based upon the results of in vitro studies. Sharma et al, for example, demonstrated increased hepatic GST expression and the attenuation of hepatotoxin-induced adduct formation following curcumin treatment[78]. Oral curcumin also led to the complete suppression of tumor NF-κB activation in an orthotopic mouse model of pancreatic cancer[94]. Anticancer activity has also been reported at a number of other sites distant from the gastrointestinal tract, including the breast[95], prostate[96], lung[97], and liver[98].
Evidence for curcumin acting synergistically/antagonistically The treatment of MCF7 breast cells with curcumin (10 µmol/L) and genistein (25 µmol/L) demonstrated a synergistic effect, leading to the total inhibition of proliferation induced by an endosulfane/chlordane/DDT mixture[99]. Curcumin also synergistically potentiated the inhibitory effect of celecoxib on pancreatic carcinoma cells[100] and additively inhibited the growth of colorectal cancer with celecoxib in the 1,2-dimethylhydrazine rat model[101].
In an in vitro model of oral cancer, EGCG blocked cells in the G0/G1 phase, while curcumin blocked in the G2/M phase of the cell cycle. The combination showed synergistic interactions in growth inhibition[102]. While tea or curcumin individually decreased the number and volume of dimethylben-zanthracene (DMBA)-induced oral tumors in hamsters, only the combination decreased the proliferation index of squamous cell carcinoma[103].
LNCaP prostate cancer cells are relatively insensitive to tumor necrosis factor related, apoptosis-inducing ligand (TRAIL). At low concentrations, neither TRAIL (20 ng/mL) nor curcumin (10 µmol/L) produced significant cytotoxicity, whereas cell death was markedly enhanced by the combina-tion. Both agents together induced the cleavage of procas-pases-8, -9, and -3, the truncation of Bid, the release of cytochrome c, and apoptosis[104].
Recent studies have also demonstrated promising interactions between curcumin and established chemotherapeutic agents. In colorectal carcinoma lines, the antiproliferative and pro-apoptotic effects of curcumin and oxaliplatin increased markedly when cells were treated with both agents[84,105]. Similarly, curcumin potentiated the pro-apoptotic effects of gemcitabine and paclitaxel in bladder cancer cell lines[106] and the antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer. Antagonistic interactions have also been demonstrated, however, with curcumin shown to inhibit chemotherapy-induced apoptosis in breast tumor lines. Camptothecin-, mechlorethamine-, and doxorubicin-induced apoptosis in MCF7, MDA-MB231 and BT474 cells was inhibited by as much as 70%, following 3 h exposure to as little as 1 μmol/L curcumin. The inhibition of both c-jun N-terminal kinase (JNK) activation and cytochrome c release occurred[107]. The same authors, using an in vivo xenograft model, found dietary curcumin (25 g/kg) decreased the level of cyclophosphamide-induced tumor regression, again with decreased JNK activation and less apoptosis.
Epigallocatechin-3-gallate (EGCG)
Green tea and its constituent molecules, including EGCG, have been found to prevent tumor formation in a wide range of tissues in animal models. However, the possible influence of green tea on cancer in humans has been difficult to interpret due to confounding factors, such as diversity in types of tea used, preparation methods, including temperature of infusion, and frequency of tea drinking.
The relevance of in vitro studies with EGCG has been reviewed by Lambert and Yang[108], who concluded that the effectiveness of tea consumption in cancer prevention remained unclear and required a better understanding of bioavailability and fundamental mechanisms.
Bioavailability of EGCG in humans A number of studies have reported the bioavailability of EGCG in various human body fluids (Table 8) following the administration of green tea or EGCG. Levels in plasma up to a maximum of 7.3 µmol/L (±3.6) have been reported, but more often are in the submicro-molar range. Bioavailability in two early studies found plasma levels at 0.2%–2% of the ingested amount of EGCG (up to 4 µmol/L), but higher plasma concentrations have since been reported in fasting patients compared to those who consumed catechins with food[109–111]. The oral administration in human patients resulted in high plasma clearance levels and volume distribution, suggesting that the bioavailability of EGCG in the blood may be low, similar to the situation found in rodents[109,112]. Dvorakova et al suggested that topical application to skin of an ointment containing 10% EGCG was likely to result in substantial intradermal uptake, but very poor systemic absorption[113].
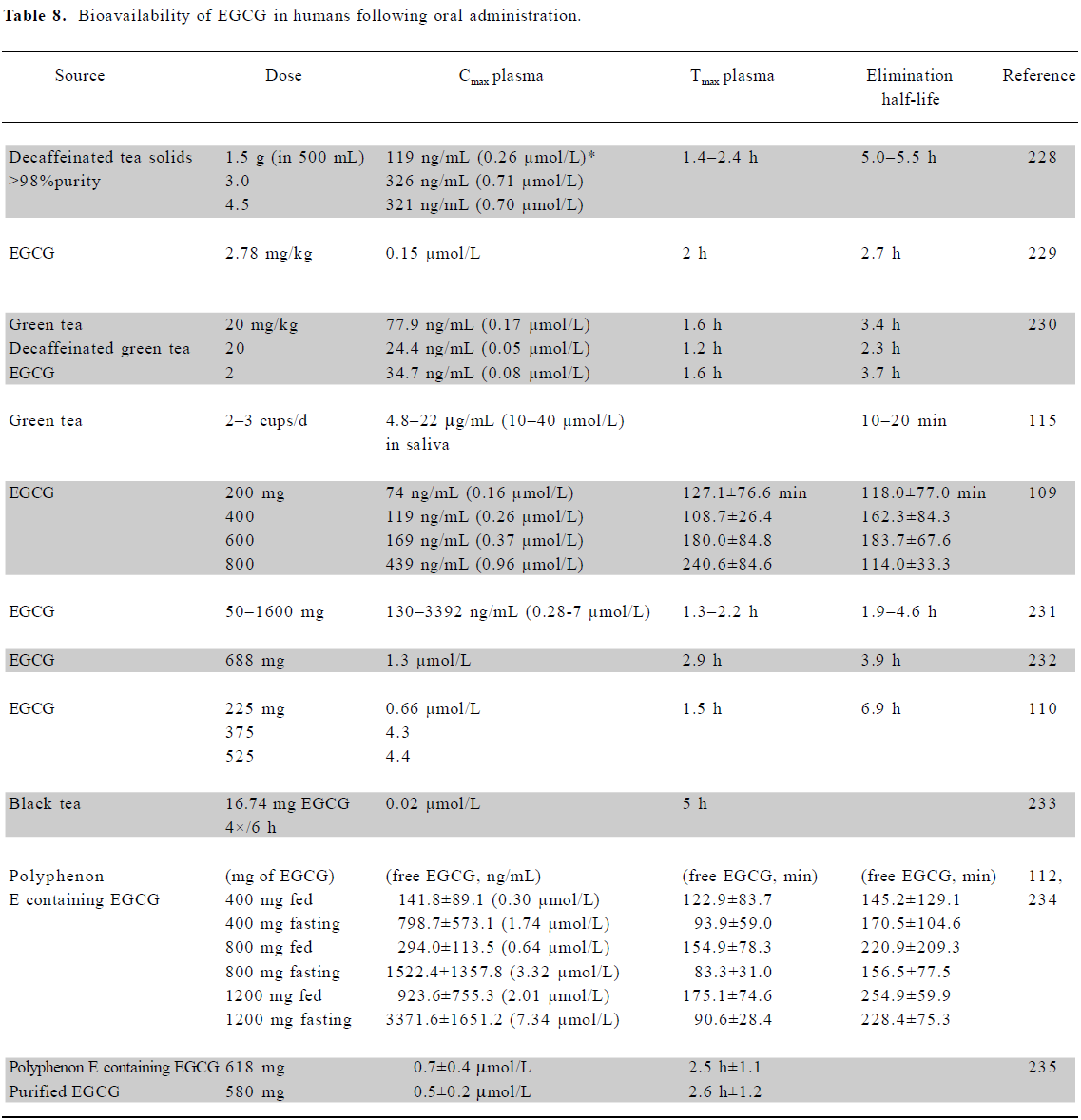
Full table
It has been found that holding a green tea solution (1.2 g/200 mL water) in the mouth for 1 min resulted in salivary EGCG concentrations (mean) of 27 µmol/L, with values up to 48 µmol/L recorded, several fold higher than that achieved through normal drinking, and many more times greater than plasma concentrations[114,115]. However, holding the tea in the mouth for 5 min resulted in salivary concentrations 4–5 times higher, whilst taking tea solids in capsules resulted in no detectable salivary catechin levels. Thus, drinking tea slowly may be an effective way of delivering relatively high concentrations to the oral cavity and esophagus.
EGCG can undergo metabolism through glucuronidation, sulfation, methylation, or ring fission[108], processes which are subject to interindividual variation.
Bioavailability of EGCG in animals Surprisingly, few studies have documented the bioavailability of EGCG in animals. Most have shown a maximum plasma concentration in the nmol/L to low µmol/L range, similar to the human situation, although 1 study using a large dose of EGCG in rats reported plasma concentrations up to 20 µmol/L (Table 9). No animal studies have examined the effect of fasting on bioavailability. Fang et al, using liposomes for the local (injection) delivery of EGCG, found that a liposomal cocktail containing deoxycholic acid and ethanol greatly increased the tumor uptake of EGCG in both melanoma and colon murine tumor models[116,117]. However, liposomal delivery was not superior following topical application.
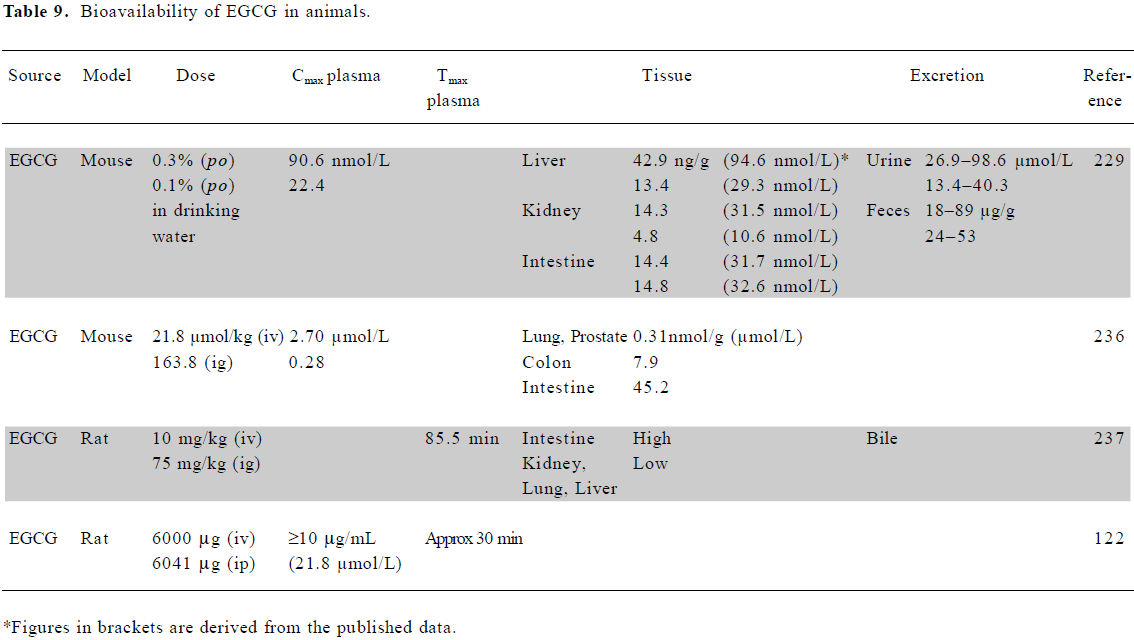
Full table
Lambert and colleagues[118] reported that piperine from black pepper enhanced the bioavailability of EGCG in mice. Small intestinal levels following EGCG administration alone resulted in a Cmax of 37.5±2.5 nmol/g at 60 min, decreasing to 5.1±1.7 nmol/g at 90 min. Following cotreatment with piperine, the Cmax was 31.6±15.1 nmol/g at 90 min, with levels still above 20 nmol/g at 180 min. The appearance of EGCG in the colon and feces was slower in the cotreated mice.
In rats and mice 24 h after the intragastric administration of radiolabeled EGCG, 10% of the dose was present in the blood, with around 1% in tissues, such as liver, kidney, heart, lung, and prostate[119]. Major elimination occurred in the feces. In line with these findings, another study suggested that the transporter-mediated intestinal efflux of catechins may play a role in the systemic elimination of these compounds[120]. Following an intravenous (iv) dose in rats, >70% was eliminated in bile and 2% in urine[119,121]. A study in rats, in which different green tea catechins were administered by iv or ip, suggested that first-pass hepatic elimination did not play a major role in the metabolism of orally-administered epigallocatechin, epicatechin, and EGCG[122].
Physiologically relevant concentrations of EGCG The plasma bioavailability of EGCG, whether administered as tea or a pure compound, is in the range of 0.1–7 µmol/L in humans, with concentrations over 100 µmol/L observed in saliva. No significant excretion occurred in urine (generally <0.1% of dose). Rodent data indicate levels up to 20 µmol/L may be achievable. Based on these data, we chose 20 µmol/L as the maximum concentration at which to consider in vitro findings.
In vitro mechanistic studies using low doses of EGCG Many in vitro studies show that EGCG, at concentrations ≤20 μmol/L, inhibits growth and induces cell cycle arrest or apoptosis in a variety of cell types (Table 10). A wide range of signaling molecules is affected, including growth factor receptors [EGFR, platelet derived (PDGFR), fibroblast (FGFR), vascular endothelial (VEGFR)], survival signaling pathway components [extracellular signal regulated kinase (ERK), p38, activating protein-1 (AP-1), signal transducer and activator of transcription (STAT), PI3K, Akt, and NF-κB], cell cycle regulators [cyclin D1, p21, p27, phosphorylated retinoblastoma (pRb), cyclin-dependent kinases (cdk)2/4/6], and regulators of apoptosis [Bcl2, Bax, Bad, caspases-3/7/8/9, and poly (ADP ribose) polymerase (PARP)]. One interesting effect of EGCG at the lowest doses (0.01–0.1 µmol/L) is the inhibition of VEGF-dependent phosphorylation of the VEGFR (Table 10), an anti-angiogenesis effect which also occurs in vivo, as discussed later.
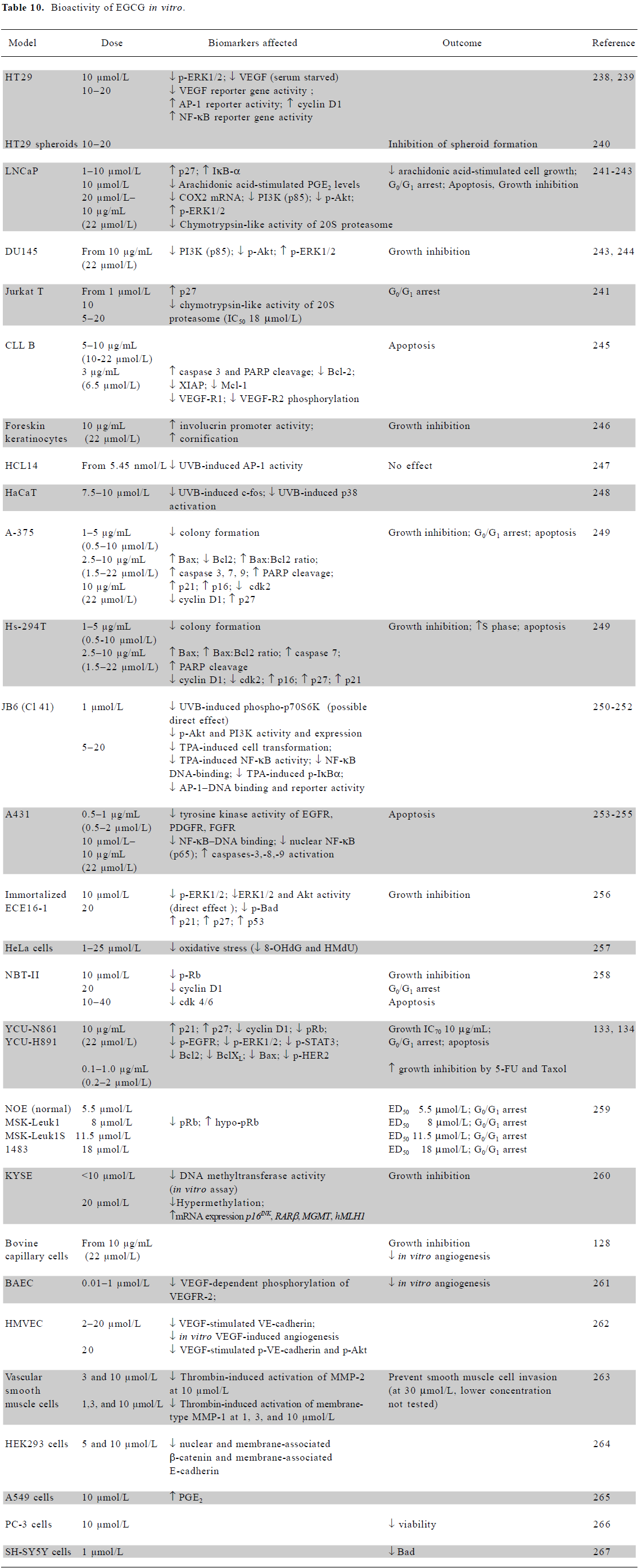
Full table
In vitro effects of EGCG observed in vivo There is substantial evidence that the effects of EGCG or green tea recorded in vitro have also been observed in animal models or humans. Green tea polyphenols inhibited the growth of 4T1 breast cancer cells and their metastasis to lungs in BALB/c mice. A reduction in tumor weight, increased survival time, and later tumor appearance were observed. The ratio of Bax/Bcl2 was altered in favor of apoptosis, along with a decrease in proliferating cell nuclear antigen and the activation of caspase-3[123]. The topical application of EGCG to SKH-1 hairless mice that had been pretreated twice weekly with UVB light decreased the multiplicity of skin tumors by 44%–72% and increased the apoptotic index by 56%–92%, again measured by increased caspase-3 activity[124]. Fang and colleagues, who demonstrated that the liposomal delivery of EGCG resulted in increased tumor uptake, also found that this delivery system led to increased antiproliferative activity in basal carcinoma cells in vitro, where the EGCG concentration in the liposomes was 21.3 μmol/L[117].
A study investigating the effect of EGCG in murine colon 26-L5 cells found that, using in vitro assays, 1,1-diphenyl-2-picryl-hydrazyl free radicals were reduced with an ED50 of 2.9 µmol/L, and cell growth was inhibited with an IC50 of 41.8 µmol/L. Following the injection of these colon cells into female BALB/c mice to analyze the effect on lung metastases, they found that EGCG, administered ip, reduced the number of tumor nodules in a dose-dependent manner[125].
When green tea extract (400–500 mg/cup, 5 cups/d) was administered for 4 weeks to 3 heavy smokers[126], smoking-induced DNA damage was decreased, cell growth (keratino-cytes) was inhibited, and the percentage of cells in S phase was reduced, with accumulation in the G0/G1 phase. DNA content became less aneuploid and p53 and caspase-3 were increased. Li and colleagues found that in hamsters, 0.6% green tea inhibited DMBA-induced oral tumor number and volume, with increased apoptosis and a decreased proliferation index and microvessel density[103]. In vitro EGCG inhibits AP-1 transcriptional activation, and this was also observed in vivo in UVB-treated transgenic mice carrying a luciferase reporter gene with an AP-1 binding sequence[127].
EGCG (10 or 50 µg/mL) significantly decreased the proliferation of bovine capillary endothelial cells, and at 1–100 µg per disc, it also inhibited neovascularization in the chick chorioallantoic membrane assay[128]. These authors also showed that green tea in drinking water (1.25% containing 708 µg/mL EGCG, giving plasma levels of 0.1–0.3 µmol/L) could significantly suppress VEGF-induced corneal neovascularization. Such results suggest that EGCG may be a useful in vivo inhibitor of angiogenesis.
Green tea consumption in two study groups, one in China and one in the USA, decreased oxidative DNA damage (8-hydroxy-deoxyguanosine in white blood cells and urine), lipid peroxidation (malondialdehyde in urine), and free radical generation (2,3-dihydroxy benzoic acid in urine) in smokers. Non-smokers (USA group) also exhibited a decrease in overall oxidative stress, which was correlated to decreased levels of free radicals[129].
A recent clinical trial involving 60 volunteers with (premalignant) prostate intraepithelial neoplasia, conducted by Bettuzzi et al, showed that after 1 year, only 1 man (3%) in the group receiving 600 mg/d green tea compounds in (oral) capsule form, presented with cancer compared to 9 (30%) from the placebo group[130]. No significant side-effects were reported. Therefore, despite the apparent poor bioavailability of green tea catechins in many studies, they appear to have great promise as chemopreventive agents.
Evidence for EGCG acting synergistically/ antagonistically There are a number of reports documenting an enhanced chemopreventive effect when EGCG or green tea is used in combination with another chemopreventive agent or a therapeutic drug.
Suganuma et al found that epicatechin significantly enhanced the uptake of labeled EGCG into human lung PC-9 cells and suggested that whole green tea was a better preventive agent than EGCG alone[131]. In this study, the pro-apoptotic effects of EGCG were also increased by tamoxifen or sulindac. Another study using the prostate cancer cell lines PC-3, LNCaP, and CWR22Rv1 showed that while 10 μmol/L EGCG only resulted in a 12%–21% inhibition in cell viability, the addition of 10 μmol/L NS-398 (a COX2 inhibitor), resulted in a 44%–49% inhibition, greater than the additive effect of either agent alone. These results corresponded to decreases in Bcl-2, procaspases-6 and -9, phospho-p65 and peroxisome proliferator-activated receptor (PPAR)γ, and increases in Bax and PARP[132].
EGCG at 0.1 µg/mL (equivalent to serum concentrations) markedly enhanced the growth inhibitory effects of 5-fluorouracil in head and neck squamous carcinoma cells[133]. The IC50 values for 2 different cell lines were reduced by ~4-fold (sensitive line) and 45-fold (resistant line). EGCG on its own at this concentration had no effect. The same group also found that EGCG enhanced the sensitivity of HNSCC (0.1 µg/mL) and breast (1.0 µg/mL) cells to Taxol[134].
Min/+ mice treated with a combination of white tea (1.5%) and sulindac (80 ppm) had significantly fewer intestinal tumors than mice treated with either agent alone. While β-catenin and β-catenin/T cell factor-4-regulated genes, cyclin D1, and c-jun were detected in polyps, the expression of these proteins was markedly reduced in the normal intestine[135]. A combination of EGCG and sulindac was also found to be efficacious in preventing azoxymethane-induced colon cancer in rat, where the combination synergistically enhanced apoptosis[136].
Resveratrol
The phytoalexin resveratrol is found largely in grape products and peanuts, with red wine a major source of human consumption. Its potential role in disease prevention is well documented, as it exhibits vasorelaxing, anti-inflammatory, antilipidemic, anti-estrogenic, antioxidant, antifungal, and antibacterial properties[137–139].
Whilst resveratrol appears to have great potential in vitro, the relevance to in vivo effects in both humans and animals is less clear, as its chemopreventive effect in vivo depends greatly on its absorption, metabolism, and tissue distribution.
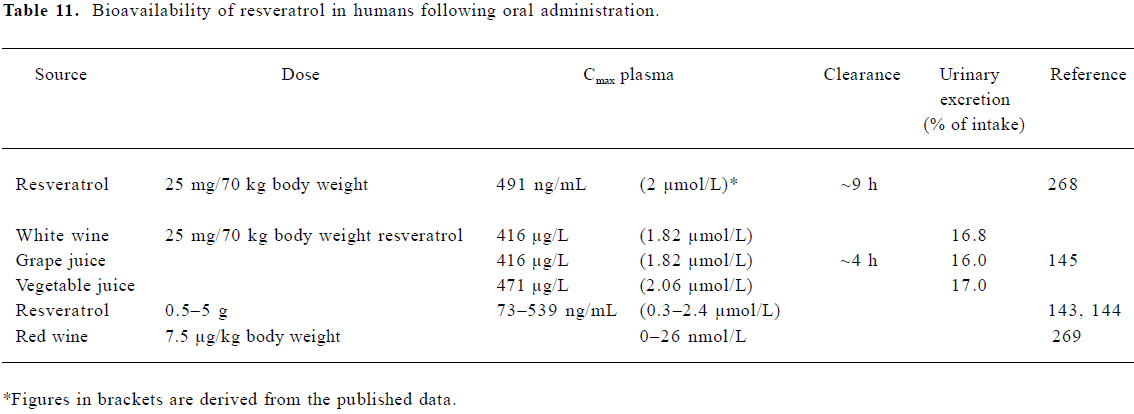
Bioavailability of resveratrol in humans Several studies have looked at the rate of uptake in healthy human volunteers via the oral administration of either resveratrol in its pure form or when present in foodstuffs (Table 11). Estimates of the amount of resveratrol in red wine (mainly trans), vary from 0.3 to 10.6 mg/L (1.3–46 µmol/L)[140–142]. Recent studies by Boocock et al[143,144] found peak resveratrol plasma concentrations of 0.3–2.4 µmol/L following single oral doses of 0.5–5 g in healthy human volunteers. Observed peak plasma concentrations for resveratrol metabolites ranged between 0.92 and 4.3 µmol/L (mono-glucuronides), and 3.7–14 µmol/L (resveratrol 3-sulfate). Plasma half-lives for the parent compound and major conjugates were of a similar order (2.9–10.6 h).
Subjects receiving a lower dose of 10–25 mg pure resvera-trol/70 kg body mass were similarly found to have serum resveratrol levels between 1.83 and 2.06 µmol/L at 30 min post-dose, returning to baseline by 4 h[145]. Wang et al[146] found both resveratrol and resveratrol glucuronide present at up to 6 h following a dose of 1 g. It is likely that conjugated resveratrol is the main component in the circulation[147], and plasma concentrations around 2 µmol/L appear typical (Table 11).
Several major metabolites of resveratrol are found in human subjects, including the sulfate-glucuronide, mono-glucuronides, and mono- and di-sulfates[143,144], with sulfation thought to occur primarily via the sulfotransferase 1A1[148] and glucuronidation via the UDP-glucuronosyltransferases[149]. The rate of glucuronidation in the human liver ranges between 0.23 and 1.2 nmol/min/mg and the rate of sulfation is 80 pmol/min per mg[141,150]. The presence of sulfated products in vivo may vary depending upon whether pure resveratrol is administered, as quercetin (also present in red wine) is known to inhibit its sulfation. Resveratrol glucuronide has been assumed to be pharmacologically inactive, although it has been suggested that glycosylation of polyphenols is an important step in protecting them from enzymatic oxidation, so extending their half-life and biological properties[151]. However, it is possible that the glucuronide may be converted back to resveratrol in vivo by the action of beta-glucuronidases[152]. The aqueous solubility of resveratrol is low, and it is thought that albumin is the main carrier in plasma, with little free resveratrol[153]. The presence of fatty acids increases binding to serum albumin, which may have important consequences for the delivery of resveratrol to cell membranes and thus signaling events.
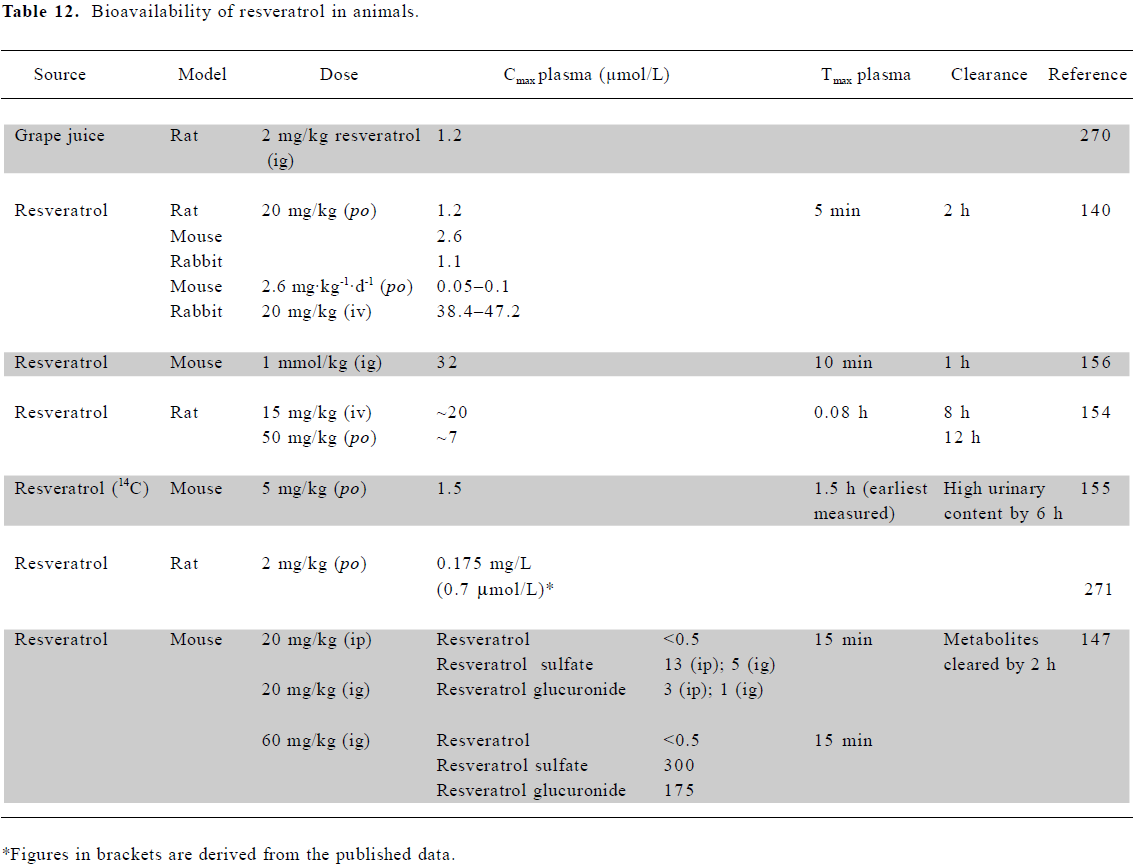
Bioavailability of resveratrol in animals Marier et al[154] observed that resveratrol was absorbed within minutes following an oral dose of 50 mg/kg to rats. Resveratrol aglycone (parent compound) plasma concentrations dropped from 10 µmol/L to levels at the limit of detection by 12 h. The glucuronide, however, was present at ~100 µmol/L, falling to 3 µmol/L after 12 h, again suggesting that resveratrol is absorbed from the intestine mainly in this form. A lower dose of 20 mg/kg gave a maximal resveratrol plasma content of 3 μmol/L, falling to <0.1 µmol/L after 1 h[140], with a 5 mg/kg dose showing maximal plasma levels of 1.5 µmol/L [155].
The disposition of resveratrol revealed far higher levels in tissues involved in absorption and excretion, such as the intestine, liver, and kidney, than in plasma. In mice, the highest accumulation was in intestinal mucosa[156], brain[157], kidneys, and liver, reaching 25–30 µmol/L following a dose of 5 mg/kg[155]. The major tissue metabolites were resveratrol-3,4'-disulfate, -3,4',5-trisulfate and β-D-glucuronide[158]. Following a 20 mg/kg dose, the rat lung contained 0.8 nmol/g resveratrol, the mouse liver 1.03 nmol/g, and the mouse kidney 0.17 nmol/g[140].
Physiologically relevant concentrations of resveratrol Animal and human studies consistently indicate resveratrol levels of 1–2 µmol/L in plasma (Tables 11, 12). Concentrations 10–20 times higher than this have been achieved through ig or iv dosing in animal studies. An examination of tissue distribution revealed that concentrations in some tissues can be significantly higher than those in plasma. Therefore, in considering the relevance of in vitro studies, we have focused on those which have reported effects at concentrations of 10 µmol/L or less.
Full table
Full table
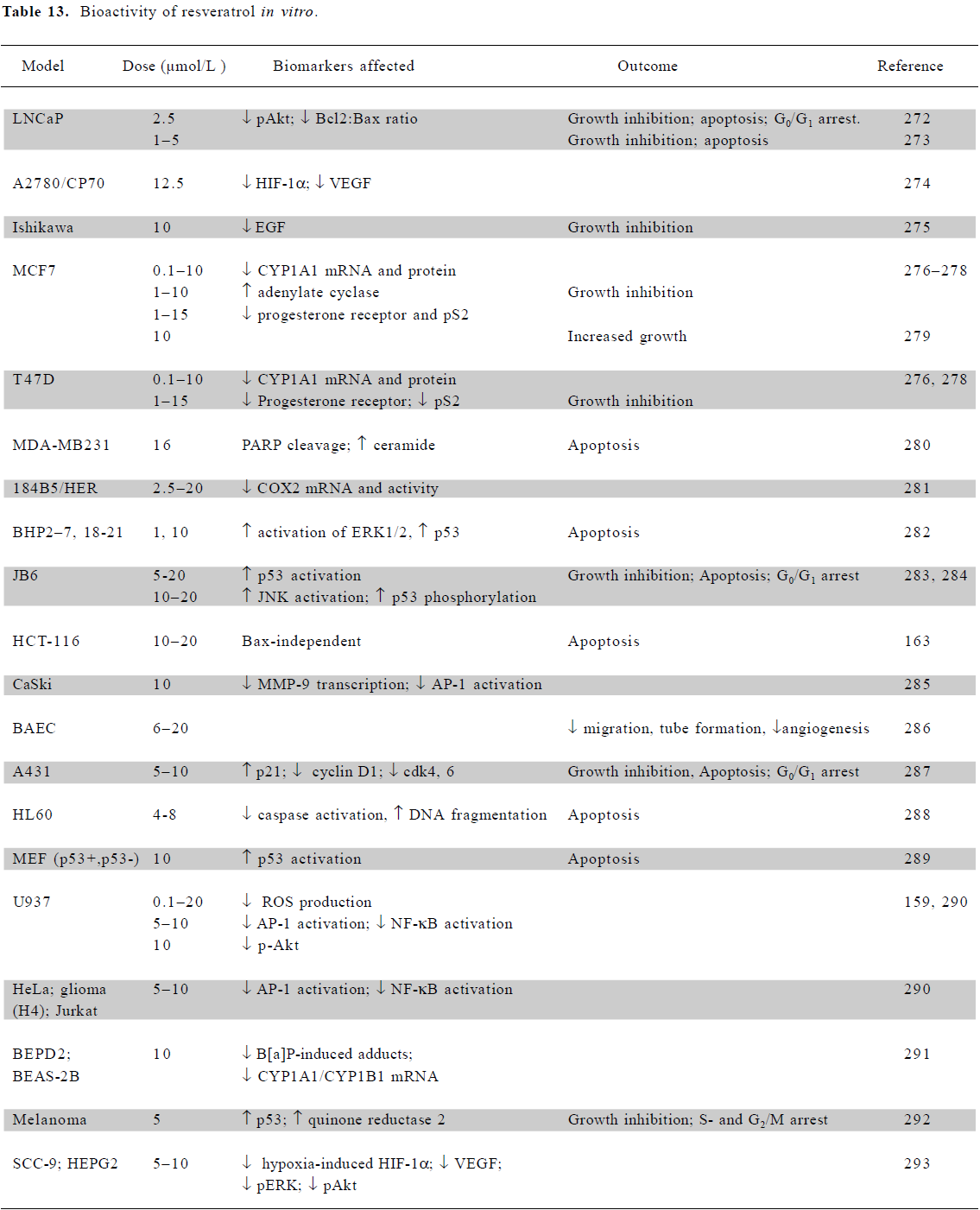
In vitro mechanistic studies using low doses of resveratrol Cell culture studies with resveratrol indicate anticancer potential over a range of doses and in a wide variety of tissues, including breast, colon, pancreas, stomach, prostate, head and neck, ovary, liver, lung, and cervix[137] (Table 13). At physiological concentrations of 10 µmol/L or less, resveratrol exhibits a range of activities which modulate signal transduction. These include the downregulation of growth factors (EGF and VEGF), alterations to survival signaling (ERK, JNK, AP-1, and NF-κB), cell cycle regulators (cyclinD1, cdk4/6, p21, and p53) and apoptosis regulators (PARP, ceramide, and caspases). In U937 monocytes, concentrations as low as 0.1 µmol/L effectively inhibited the production of ROS, with the inhibition of Akt phosphorylation at 10 µmol/L [159].
Full table
In vitro effects of resveratrol observed in vivo Whether or not the IC50 values in vitro are achievable in vivo depends to some extent on the target tissue. It is likely that the highest concentration of resveratrol and its metabolites following ingestion would be achieved in colorectal mucosal tissue and in the liver. Several studies administering resveratrol via ingestion of red wine have used the mean serum antioxidant capacity as a marker of efficacy. In healthy volunteers consuming 300 mL red wine over a 30 min period, blood taken up to 2 h post-dose showed significantly raised serum antioxidant capacity[160].
MDA-MB231 xenografts in nude mice exhibited an increase in the apoptotic index and decreased angiogenesis when treated daily with 25 mg/kg resveratrol for 3 weeks, while the same cell line in the culture did not undergo apoptosis at concentrations less than 100 µmol/L[161]. Conversely, when B16M tumor cells were inoculated into mice, 20 mg/kg resveratrol did not affect tumor growth (tumor concentration of 0.04 nmol/g), even though in the culture the cells underwent 60% apoptosis following a 5 µmol/L treatment for 24 h[140]. The rats inoculated with the Yoshida ascites hepatoma receiving daily ip injections (1 mg/kg) of resveratrol exhibited decreased tumor growth due to the induction of apoptosis and a G2/M cell cycle arrest. This effect was not seen in vitro using resveratrol in the range of 15–30 µmol/L over 24 h[162]. Daily ip injections of 40 mg/kg resveratrol (estimated serum level of 25 µmol/L) reduced neuroblastoma growth in rats and increased survival by 70%. In culture, resveratrol was also cytotoxic to neuroblastoma cells in a range from 10 to 100 µmol/L [163]. Resveratrol at 100 mg/kg per day prolonged the survival time for rats with intracerebral tumors generated from RT-2 glioma cell xenografts. The IC50 for RT-2 cells in the culture equated to 12.8 µmol/L following a 48 h treatment, with 39% cells undergoing apoptosis at the higher concentration of 25 µmol/L[164].
A number of reports have shown that resveratrol can inhibit NF-κB activation in vitro. Banerjee et al found that in rats, 10 ppm produced striking reductions in DMBA-induced breast tumor incidence and multiplicity, while extending tumor latency[165]. They reported that resveratrol suppressed DMBA-induced COX2 and matrix metallopro-teinase (MMP)-9 expressions through the downregulation of NF-κB activation.
Resveratrol treatment may also inhibit preneoplastic conditions. In both an experimentally induced model of colitis[166] and the Min/+ mouse[167], resveratrol was able to reduce damage/adenoma load and COX2 protein expression. The spontaneous development of mammary tumors in HER2/neu mice was delayed with the reduction in both size and number of tumors following resveratrol treatment[168]. In rats, azoxymethane treatment caused the formation of aberrant crypt foci, the number of which was significantly reduced in the presence of resveratrol (200 µg/kg per day for 100 d), with decreased bax and increased p21 expression in the crypts[169]. The treatment of dimethylhydrazine-induced aberrant crypt foci with resveratrol (8 mg/kg per day) resulted in a marked reduction in tumor incidence and degree of histological lesions[170]. Similarly, rats fed a diet containing 15% grape extract showed a decrease in the number and area of GST-P+ve foci[171].
Evidence for resveratrol acting synergistically/antagonistically Resveratrol sensitized colon cancer cells to CD95 and the TRAIL-mediated induction of apoptosis, and at 10 µmol/L, sensitized HT29 cells to cisplatin-induced apopto-sis[172]. Fulda and Debatin found that pretreatment with resveratrol cooperatively enhanced doxorubicin, cytarabine, actinomycin D, Taxol, and methotrexate-induced apoptosis and cell cycle arrest in neuroblastoma cells[173], and enhanced TRAIL-mediated apoptosis in neuroblastoma and Jurkat T cells[174]. Resveratrol (10 μmol/L) also enhanced the apoptotic effects of paclitaxel in A549, EBC-1, and Lu65 lung cancer cell lines[175], and of cisplatin and doxorubicin in OVCAR-3 and Ishikawa cells, respectively[176].
Resveratrol has been used in combination with other phytochemicals, such as betasiterol, resulting in enhanced growth inhibition due to an arrest at both the G1 and G2/M phases of the cell cycle in PC-3 cells[177]. The combination of quercetin/ellagic acid with resveratrol resulted in a synergistic effect on caspase-3 activation leading to apoptosis[178]. Lee et al[179] found individual concentrations of resveratrol (0.5 µmol/L) or I3C (50 µmol/L) to induce the non-steroidal, anti-inflammatory, drug-activated gene (NAG)-1, a TGF-β superfamily gene associated with pro-apoptotic and anti-tumorigenic activity. However, when used in combination, the doses could be reduced to 0.025 and 1 µmol/L, respectively.
Conclusion
Plasma concentrations in humans, following normal dietary intake or administration of supplements or formula-tions, have been measured or can be estimated from animal studies for each of the agents reviewed. In general, achievable plasma concentrations were in the low micromolar range, although animal studies revealed the possibility of considerably higher concentrations in some tissues. In summarizing data from many cell culture studies, which have been carried out using the low concentrations achievable in vivo, it is apparent that all the compounds still exhibit biological activity. However, the range of activities is more limited, compared to that using a much wider dose range. While this may reflect genuine lack of activity at low doses, it is partly due to the fact that many studies have chosen to use only higher doses and shorter time points. There may therefore be many more useful preventive possibilities to be identified using lower doses.
Two very encouraging themes emerge from the data reviewed here. First, while not all in vitro findings are matched in vivo, many observations have been validated in animals or humans, giving credibility to the value of cell cultures for screening and more detailed mechanistic studies. However, some caution is required in extrapolation, as in many cases it is not known whether exactly the same signaling mechanisms are operational in vivo. There can also be significant discrepancies in the effective doses, even to the extent that where low levels are active in vivo, much higher concentrations are required to achieve the same effect in vitro.
Second, there is a growing body of evidence to suggest that even if single agents are inactive at low concentrations, combinations of 2 or more compounds might be much more efficacious. Combinations with chemotherapeutic drugs also offer the possibility of lowering the dose of the latter, with a consequent reduction of unwanted side-effects.
Thus, studies in cell culture have provided valuable insights into chemopreventive mechanisms of action, and there is now a need to pursue these at physiological doses and in novel combinations. One further aspect, which has not been tackled in any detail here, is the need to address more rigorously the question of tissue specificity and cancer subtype.
References
- Manson MM. Inhibition of survival signalling by dietary polyphenols and indole-3-carbinol. Eur J Cancer 2005;41:1842-53.
- Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol 2006;71:1397-421.
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 2003;3:768-80.
- Sepkovic DW, Bradlow HL, Bell MC. Quantitative determination of 3,3'-diindolylmethane in urine of individuals receiving indole-3-carbinol. Nutr Cancer 2001;42:57-63.
- Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3'-diindolylmethane. Cancer Epidemiol Biomarkers Prev 2006;15:2477-81.
- Dalessandri KM, Firestone GL, Fitch MD, Bradlow HL, Bjeldanes LF. Pilot study: Effect of 3,3'-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr Cancer 2004;50:161-7.
- Bell MC, Crowley-Nowick P, Bradlow HL, Sepkovic DW, Schmidt-Grimminger D, Howell P, et al. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynaecol Oncol 2000;78:123-9.
- Bradlow HL, Michnovicz JJ, Halper M, Miller DG, Wong GYC, Osborne MP. Long-term responses of women to indole-3-carbinol or a high fiber diet. Cancer Epidemiol Biomarkers Prev 1994;3:591-5.
- McAlindon TE, Gulin J, Chen T, Klug T, Lahita R, Nuite M. Indole-3-carbinol in women with SLE: effect on estrogen metabolism and disease activity. Lupus 2001;10:779-83.
- Michnovicz JJ. Increased estrogen 2-hydroxylation in obese women using oral indole-3-carbinol. Int J Obes 1998;22:227-9.
- Michnovicz JJ, Adlercreutz H, Bradlow HL. Changes in levels of urinary estrogen metabolites after oral indole-3-carbinol treatment in humans. J Natl Cancer Inst 1997;89:718-23.
- Michnovicz JJ, Bradlow HL. Altered estrogen metabolism and excretion in humans following consumption of indole-3-carbinol. Nutr Cancer 1991;16:59-66.
- Rosen CA, Bryson PC. Indole-3-carbinol for recurrent respiratory papillomatosis: long-term results. J Voice 2004;18:248-53.
- Naik R, Nixon S, Lopes A, Godfrey K, Hatem MH, Monaghan JM. A randomized phase II trial of indole-3-carbinol in the treatment of vulvar intraepithelial neoplasia. Int J Gynecol Cancer 2006;16:786-90.
- Wong GYC, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl 1997;28/29:111-6.
- Reed GA, Peterson KS, Smith HJ, Gray JC, Sullivan DK, Mayo MS, et al. A phase I study of indole-3-carbinol in women: tolerability and effects. Cancer Epidemiol Biomarkers Prev 2005;14:1953-60.
- Shertzer HG, Berger ML, Tabor MW. Intervention in free-radical mediated hepatotoxicity and lipid-peroxidation by indole-3-carbinol. Biochem Pharmacol 1988;37:333-8.
- Stresser DM, Williams DE, Griffin DA, Bailey GS. Mechanisms of tumor modulation by indole-3-carbinol. Disposition and excretion in male Fischer 344 rats. Drug Metab Disp 1995;23:965-75.
- Yu Z, Mahadevan B, Lohr CV, Fischer KA, Louderback MA, Krueger SK, et al. Indole-3-carbinol in the maternal diet provides chemoprotection for the fetus against transplacental carcinogenesis by the polycyclic aromatic hydrocarbon, dibenzo[a,l]pyrene. Carcinogenesis 2006;27:2116-23.
- Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, et al. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res 2004;10:5233-41.
- Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, et al. Physiological modeling of formulated and crystalline 3,3'-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Disp 2004;32:632-8.
- Crowell JA, Page JG, Levine BS, Tomlinson MJ, Hebert CD. Indole-3-carbinol, but not its major digestive product 3,3'-diindolylmethane, induces reversible hepatocyte hypertrophy and cytochromes P450. Toxicol Appl Pharmacol 2006;211:115-23.
- Donald S, Verschoyle RD, Greaves P, Colombo T, Zucchetti M, Falcioni C, et al. Dietary agent indole-3-carbinol protects female rats against the hepatotoxicity of the antitumor drug ET-743 (trabectidin) without compromising efficacy in a rat mammary carcinoma. Int J Cancer 2004;111:961-7.
- Leibelt DA, Hedstrom OR, Fischer KA, Pereira CB, Williams DE. Evaluation of chronic dietary exposure to indole-3-carbinol and absorption-enhanced 3,3'-diindolylmethane in sprague-dawley rats. Toxicol Sci 2003;74:10-21.
- Zhang X, Malejka-Giganti D. Effects of treatment of rats with indole-3-carbinol on apoptosis in the mammary gland and mammary adenocarcinomas. Anticancer Res 2003;23:2473-79.
- Parkin DR, Malejka-Giganti D. Differences in the hepatic P450-dependent metabolism of estrogen and tamoxifen in response to treatment of rats with 3,3'-diindolylmethane and its parent compound indole-3-carbinol. Cancer Detect Prev 2004;28:72-9.
- Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 1998;19:1631-39.
- Hestermann EV, Brown M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol Cell Biol 2003;23:7920-25.
- Liu S, Abdelrahim M, Khan S, Ariazi E, Jordan VC, Safe S. Aryl hydrocarbon receptor agonists directly activate estrogen receptor alpha in MCF-7 breast cancer cells. Biol Chem 2006;387:1209-13.
- Jellinck PH, Forkert P-G, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem Pharmacol 1993;45:1129-36.
- Li Y, Li X, Sarkar FH. Gene expression profiles of I3C- and DIM-treated PC3 human prostate cancer cells determined by cDNA microarray analysis. J Nutr 2003;133:1011-19.
- Meng Q, Yuan F, Goldberg ID, Rosen EM, Auborn K, Fan S. Indole-3-carbinol is a negative regulator of estrogen receptor-a signalling in human tumor cells. J Nutr 2000;130:2927-31.
- Sundar SN, Kerekatte V, Equinozio CN, Doan VB, Bjeldanes LF, Firestone GL. Indole-3-carbinol selectively uncouples expression and activity of estrogen receptor subtypes in human breast cancer cells. Mol Endocrinol 2006;20:3070-82.
- Riby JE, Chang GHF, Firestone GL, Bjeldanes LF. Ligand-independent activation of estrogen receptor function by 3,3'-diindolylmethane in human breast cancer cells. Biochem Pharmacol 2000;60:167-77.
- Leong H, Firestone GL, Bjeldanes LF. Cytostatic effects of 3,3'-diindolylmethane in human endometrial cancer cells result from an estrogen receptor-mediated increase in transforming growth factor-α expression. Carcinogenesis 2001;22:1809-17.
- Leong H, Riby JE, Firestone GL, Bjeldanes LF. Potent ligand-independent estrogen receptor activation by 3,3'-diindolyl-methane is mediated by cross talk between the protein kinase A and mitogen-activated protein kinase signaling pathways. Mol Endocrinol 2004;18:291-302.
- Wang TT, Milner MJ, Milner JA, Kim YS. Estrogen receptor alpha as a target for indole-3-carbinol. J Nutr Biochem 2006;17:659-64.
- Le HT, Schaldach CM, Firestone GL, Bjeldanes LF. Plant-derived 3,3'-diindolylmethane is a strong androgen antagonist in human prostate cancer cells. J Biol Chem 2003; 278: 21 136–45.
- Hsu JC, Zhang J, Dev A, Wing A, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibition of androgen receptor expression and down-regulation of androgen responsiveness in human prostate cancer cells. Carcinogenesis 2005;26:1896-904.
- Moiseeva EP, Fox LH, Howells LM, Temple LA, Manson MM. Indole-3-carbinol-induced death in cancer cells involves EGFR downregulation and is exacerbated in a 3D environment. Apopto-sis 2006;11:799-812.
- Moiseeva EP, Heukers R, Manson MM. EGFR and Src are involved in indole-3-carbinol-induced death and cell cycle arrest of human breast cancer cells. Carcinogenesis 2007;28:435-45.
- Howells LM, Hudson EA, Manson MM. Inhibition of phosphati-dylinositol 3-kinase/protein kinase B signaling is not sufficient to account for indole-3-carbinol-induced apoptosis in some breast and prostate tumor cells. Clin Cancer Res 2005;11:8521-27.
- Fan S, Meng Q, Auborn K, Carter T, Rosen EM. BRCA1 and BRCA2 as molecular targets for phytochemicals indole-3-carbinol and genistein in breast and prostate cancer cells. Br J Cancer 2006;94:407-26.
- Gong Y, Firestone GL, Bjeldanes LF. 3,3'-diindolylmethane is a novel topoisomerase IIalpha catalytic inhibitor that induces S-phase retardation and mitotic delay in human hepatoma HepG2 cells. Mol Pharmacol 2006;69:1320-27.
- Gong Y, Sohn H, Xue L, Firestone GL, Bjeldanes LF. 3,3'-Diindolylmethane is a novel mitochondrial H(+)-ATP synthase inhibitor that can induce p21(Cip1/Waf1) expression by induction of oxidative stress in human breast cancer cells. Cancer Res 2006;66:4880-87.
- Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3'-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 2006;27:717-28.
- Savino JA 3rd, Evans JF, Rabinowitz D, Auborn KJ, Carter TH. Multiple, disparate roles for calcium signaling in apoptosis of human prostate and cervical cancer cells exposed to diindolyl-methane. Mol Cancer Ther 2006;5:556-63.
- Xue L, Firestone GL, Bjeldanes LF. DIM stimulates IFNγ gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene 2005;24:2343-53.
- Rahman KMW, Li Y, Sarkar FH. Inactivation of Akt and NF-κB play important roles during indole-3-carbinol-induced apoptosis in breast cancer cells. Nutr Cancer 2004;48:84-94.
- Rahman KMW, Sarkar FH. Inhibition of nuclear translocation of nuclear factor-κB contributes to 3,3'-diindolylmethane-induced apoptosis in breast cancer cells. Cancer Res 2005;65:364-71.
- Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by AKT and NF-kappaB pathways in prostate cancer cells. Front Biosci 2005;10:236-43.
- Takada Y, Andreeff M, Aggarwal BB. Indole-3-carbinol suppresses NF-kappaB and I{kappa}B{alpha} kinase activation causing inhibition of expression of NF-kappaB-regulated anti-apoptotic and metastatic gene products and enhancement of apoptosis in myeloid and leukemia cells. Blood 2005;106:641-9.
- Bhuiyan MM, Li Y, Banerjee S, Ahmed F, Wang Z, Ali S, . Down-regulation of androgen receptor by 3,3'-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells. Cancer Res 2006; 66: 10 064–72.
- IARC. Handbooks of cancer prevention; N 9. Cruciferous vegetables, isothiocyanates and indoles. Lyon: IARC Press; 2004.
- Grubbs CJ, Steele VE, Casebolt T, Juliana MM, Eto I, Whitaker LM, et al. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res 1995;15:709-16.
- Chen DZ, Qi M, Auborn KJ, Carter TH. Indole-3-carbinol and diindolylmethane induce apoptosis of human cervical cancer cells and in murine HPV-16 transgenic preneoplastic cervical epithelium. J Nutr 2001;131:3294-302.
- Bradlow HL, Michnovicz JJ, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis 1991;12:1571-74.
- McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res 2001;61:3299-307.
- Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 2002;365:405-16.
- Katchamart S, Stresser DM, Dehal SS, Kupfer D, Williams DE. Concurrent flavin-containing monooxygenase down-regulation and cytochrome P-450 induction by dietary indoles in rat: implications for drug-drug interaction. Drug Metab Disp 2000;28:930-6.
- Katchamart S, Williams DE. Indole-3-carbinol modulation of hepatic monooxygenases CYP1A1, CYP1A2 and FMO1 in guinea pig, mouse and rabbit. Comp Biochem Physiol Part C 2001;129:377-84.
- Manson MM, Hudson EA, Ball HWL, Barrett MC, Clark HL, Judah DJ, et al. Chemoprevention of aflatoxin B1-induced carcinogenesis by indole-3-carbinol in rat liver - predicting the outcome using early biomarkers. Carcinogenesis 1998;19:1829-36.
- Park J-Y, Bjeldanes LF. Organ-selective induction of cytochrome P-450-dependent activities by indole-3-carbinol-derived products: influence on covalent binding of benzo[a]pyrene to hepatic and pulmonary DNA in the rat. Chem Biol Interact 1992;83:235-47.
- Hudson EA, Howells LM, Gallacher-Horley B, Fox LH, Gescher A, Manson MM. Growth-inhibitory effects of the chemopreven-tive agent indole-3-carbinol are increased in combination with the polyamine putrescine in the SW480 colon tumour cell line. BMC Cancer 2003;3:2.
- Rahman KM, Sarkar FH, Banerjee S, Wang Z, Liao DJ, Hong X, et al. Therapeutic intervention of experimental breast cancer bone metastasis by indole-3-carbinol in SCID-human mouse model. Mol Cancer Ther 2006;5:2747-56.
- Arora A, Shukla Y. Modulation of vinca-alkaloid induced P-glycoprotein expression by indole-3-carbinol. Cancer Lett 2003;189:167-73.
- Arora A, Seth K, Kalra N, Shukla Y. Modulation of P-glycoprotein-mediated multidrug-resistance in K562 leukemic cells by indole-3-carbinol. Toxicol Appl Pharmacol 2005;202:237-43.
- Christensen JG, LeBlanc GA. Reversal of multidrug resistance in vivo by dietary administration of the phytochemical indole-3-carbinol. Cancer Res 1996;56:574-81.
- Auborn KJ, Fan S, Rosen EM, Goodwin L, Chandraskaren A, Williams DE, et al. Indole-3-carbinol is a negative regulator of estrogen. J Nutr 2003;133:S2470-5.
- Cover CM, Hsieh SJ, Cram EJ, Hong C, Riby JE, Bjeldanes LF, et al. Indole-3-carbinol and tamoxifen cooperate to arrest the cell cycle of MCF-7 human breast cancer cells. Cancer Res 1999;59:1244-51.
- Malejka-Giganti D, Parkin DR, Bennett KK, Lu Y, Decker RW, Niehans GA, et al. Suppression of mammary gland carcinogenesis by post-initiation treatment of rats with tamoxifen or indole-3-carbinol or their combination. Eur J Cancer Prev 2007;16:130-41.
- Nho CW, Jeffery E. The synergistic upregulation of phase II detoxification enzymes by glucosinolate breakdown products in cruciferous vegetables. Toxicol Appl Pharmacol 2001;174:146-52.
- Ireson C, Orr S, Jones DJ, Verschoyle R, Lim CK, Luo JL, et al. Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res 2001;61:1058-64.
- Ireson CR, Jones DJ, Orr S, Coughtrie MW, Boocock DJ, Williams ML, et al. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomarkers Prev 2002;11:105-11.
- Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001;21:2895-900.
- Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res 2004;10:6847-54.
- Garcea G, Berry DP, Jones DJ, Singh R, Dennison AR, Farmer PB, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev 2005;14:120-5.
- Sharma RA, Ireson CR, Verschoyle RD, Hill KA, Williams ML, Leuratti C, et al. Effects of dietary curcumin on glutathione S-transferase and malondialdehyde-DNA adducts in rat liver and colon mucosa: relationship with drug levels. Clin Cancer Res 2001;7:1452-8.
- Perkins S, Verschoyle RD, Hill K, Parveen I, Threadgill MD, Sharma RA, et al. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev 2002;11:535-40.
- Ravindranath V, Chandrasekhara N. Absorption and tissue distribution of curcumin in rats. Toxicol 1980;16:259-65.
- Marczylo TH, Verschoyle RD, Cooke DN, Morazzoni P, Steward WP, Gescher AJ. Comparison of systemic availability of curcumin with that of curcumin formulated with phosphatidyl-choline. Cancer Chemother Pharmacol 2006;60:171-7.
- Liu A, Lou H, Zhao L, Fan P. Validated LC/MS/MS assay for curcumin and tetrahydrocurcumin in rat plasma and application to pharmacokinetic study of phospholipid complex of curcumin. J Pharm Biomed Anal 2006;40:720-7.
- Maiti K, Mukherjee K, Gantait A, Saha BP, Mukherjee PK. Curcumin-phospholipid complex: Preparation, therapeutic evaluation and pharmacokinetic study in rats. Int J Pharm 2007;330:155-63.
- Li L, Ahmed B, Mehta K, Kurzrock R. Liposomal curcumin with and without oxaliplatin: effects on cell growth, apoptosis, and angiogenesis in colorectal cancer. Mol Cancer Ther 2007;6:1276-82.
- Li L, Braiteh FS, Kurzrock R. Liposome-encapsulated curcumin: in vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer 2005;104:1322-31.
- Bisht S, Feldmann G, Soni S, Ravi R, Karikari C, Maitra A, et al. Polymeric nanoparticle-encapsulated curcumin (nanocurcumin): a novel strategy for human cancer therapy. J Nanobiotechnol 2007;5:3.
- Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 2003;23:363-98.
- Elattar TM, Virji AS. The inhibitory effect of curcumin, genistein, quercetin and cisplatin on the growth of oral cancer cells in vitro. Anticancer Res 2000;20:1733-8.
- Squires MS, Hudson EA, Howells L, Sale S, Houghton CE, Jones JL, et al. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochem Pharmacol 2003;65:361-76.
- Samaha HS, Kelloff GJ, Steele V, Rao CV, Reddy BS. Modulation of apoptosis by sulindac, curcumin, phenylethyl-3-methylcaffeate, and 6-phenylhexyl isothiocyanate: apoptotic index as a biomarker in colon cancer chemoprevention and promotion. Cancer Res 1997;57:1301-5.
- Rao CV, Rivenson A, Simi B, Reddy BS. Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally occurring plant phenolic compound. Cancer Res 1995;55:259-66.
- Mahmoud NN, Carothers AM, Grunberger D, Bilinski RT, Churchill MR, Martucci C, et al. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis 2000;21:921-7.
- Chuang SE, Cheng AL, Lin JK, Kuo ML. Inhibition by curcumin of diethylnitrosamine-induced hepatic hyperplasia, inflamma-tion, cellular gene products and cell-cycle-related proteins in rats. Food Chem Toxicol 2000;38:991-5.
- Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-êB-regulated gene products. Cancer Res 2007;67:3853-61.
- Inano H, Onoda M. Prevention of radiation-induced mammary tumors. Int J Radiat Oncol Biol Phys 2002;52:212-23.
- Frank N, Knauft J, Amelung F, Nair J, Wesch H, Bartsch H. No prevention of liver and kidney tumors in Long-Evans Cinnamon rats by dietary curcumin, but inhibition at other sites and of metastases. Mutat Res 2003;523-524:127-35.
- Menon LG, Kuttan R, Kuttan G. Inhibition of lung metastasis in mice induced by B16F10 melanoma cells by polyphenolic compounds. Cancer Lett 1995;95:221-5.
- Busquets S, Carbo N, Almendro V, Quiles MT, Lopez-Soriano FJ, Argiles JM. Curcumin, a natural product present in turmeric, decreases tumor growth but does not behave as an anticachectic compound in a rat model. Cancer Lett 2001;167:33-8.
- Verma SP, Salamone E, Goldin B. Curcumin and genistein, plant natural products, show synergistic inhibitory effects on the growth of human breast cancer MCF-7 cells induced by estrogenic pesticides. Biochem Biophys Res Commun 1997;233:692-6.
- Lev-Ari S, Zinger H, Kazanov D, Yona D, Ben-Yosef R, Starr A, et al. Curcumin synergistically potentiates the growth inhibitory and pro-apoptotic effects of celecoxib in pancreatic adenocarcinoma cells. Biomed Pharmacother 2005;59 Suppl 2:S276-80.
- Lev-Ari S, Strier L, Kazanov D, Madar-Shapiro L, Dvory-Sobol H, Pinchuk I, et al. Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin Cancer Res 2005;11:6738-44.
- Khafif A, Schantz SP, Chou TC, Edelstein D, Sacks PG. Quantitation of chemopreventive synergism between (-)-epigallocatechin-3-gallate and curcumin in normal, premalignant and malignant human oral epithelial cells. Carcinogenesis 1998;19:419-24.
- Li N, Chen X, Liao J, Yang G, Wang S, Josephson Y, et al. Inhibition of 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamsters by tea and curcumin. Carcinogenesis 2002;23:1307-13.
- Deeb D, Xu YX, Jiang H, Gao X, Janakiraman N, Chapman RA, et al. Curcumin (diferuloyl-methane) enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in LNCaP prostate cancer cells. Mol Cancer Ther 2003;2:95-103.
- Howells LM, Mitra A, Manson MM. Comparison of oxaliplatin- and curcumin-mediated antiproliferative effects in colorectal cell lines. Int J Cancer 2007;121:175-83.
- Kamat AM, Sethi G, Aggarwal BB. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor-kappaB and nuclear factor-kappaB-regulated gene products in IFN-alpha-sensitive and IFN-alpha-resistant human bladder cancer cells. Mol Cancer Ther 2007;6:1022-30.
- Somasundaram S, Edmund NA, Moore DT, Small GW, Shi YY, Orlowski RZ. Dietary curcumin inhibits chemotherapy-induced apoptosis in models of human breast cancer. Cancer Res 2002;62:3868-75.
- Lambert JD, Yang CS. Mechanisms of cancer prevention by tea constituents. J Nutr 2003;133:S3262-7.
- Chow HH, Cai Y, Alberts DS, Hakim I, Dorr R, Shahi F, et al. Phase I pharmacokinetic study of tea polyphenols following single-dose administration of epigallocatechin gallate and polyphenon E. Cancer Epidemiol Biomarkers Prev 2001;10:53-8.
- Nakagawa K, Okuda S, Miyazawa T. Dose-dependent incorporation of tea catechins, (-)-epigallocatechin-3-gallate and (-)-epigallocatechin, into human plasma. Biosci Biotechnol Biochem 1997;61:1981-5.
- Unno T, Kondo K, Itakura H, Takeo T. Analysis of (-)-epigallocatechin gallate in human serum obtained after ingesting green tea. Biosci Biotechnol Biochem 1996;60:2066-68.
- Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res 2003;9:3312-9.
- Dvorakova K, Dorr RT, Valcic S, Timmermann B, Alberts DS. Pharmacokinetics of the green tea derivative, EGCG, by the topical route of administration in mouse and human skin. Cancer Chemother Pharmacol 1999;43:331-5.
- Yang CS, Lee MJ, Chen L. Human salivary tea catechin levels and catechin esterase activities: implication in human cancer prevention studies. Cancer Epidemiol Biomarkers Prev 1999;8:83-9.
- Yang CS, Kim S, Yang GY, Lee MJ, Liao J, Chung JY, et al. Inhibition of carcinogenesis by tea: bioavailability of tea poly-phenols and mechanisms of actions. Proc Soc Exp Biol Med 1999;220:213-7.
- Fang JY, Hung CF, Hwang TL, Huang YL. Physicochemical characteristics and in vivo deposition of liposome-encapsulated tea catechins by topical and intratumor administrations. J Drug Target 2005;13:19-27.
- Fang JY, Lee WR, Shen SC, Huang YL. Effect of liposome encapsulation of tea catechins on their accumulation in basal cell carcinomas. J Dermatol Sci 2006;42:101-9.
- Lambert JD, Hong J, Kim DH, Mishin VM, Yang CS. Piperine enhances the bioavailability of the tea polyphenol (–)-epigallo-catechin-3-gallate in mice. J Nutr 2004;134:1948-52.
- Kohri T, Nanjo F, Suzuki M, Seto R, Matsumoto N, Yamakawa M, et al. Synthesis of (-)-[4-3H]epigallocatechin gallate and its metabolic fate in rats after intravenous administration. J Agric Food Chem 2001;49:1042-8.
- Vaidyanathan JB, Walle T. Transport and metabolism of the tea flavonoid (–)-epicatechin by the human intestinal cell line Caco-2. Pharm Res 2001;18:1420-5.
- Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H, Fujiki H. Wide distribution of [3H](–)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 1998;19:1771-6.
- Cai Y, Anavy ND, Chow HH. Contribution of presystemic hepatic extraction to the low oral bioavailability of green tea catechins in rats. Drug Metab Dispos 2002;30:1246-9.
- Baliga MS, Meleth S, Katiyar SK. Growth inhibitory and anti-metastatic effect of green tea polyphenols on metastasis-specific mouse mammary carcinoma 4T1 cells in vitro and in vivo systems. Clin Cancer Res 2005;11:1918-27.
- Lu YP, Lou YR, Xie JG, Peng QY, Liao J, Yang CS, . Topical applications of caffeine or (–)-epigallocatechin gallate (EGCG) inhibit carcinogenesis and selectively increase apoptosis in UVB-induced skin tumors in mice. Proc Natl Acad Sci USA 2002; 99: 12 455–60.
- Ogasawara M, Matsunaga T, Suzuki H. Differential effects of antioxidants on the in vitro invasion, growth and lung metastasis of murine colon cancer cells. Biol Pharm Bull 2007;30:200-4.
- Schwartz JL, Baker V, Larios E, Chung FL. Molecular and cellular effects of green tea on oral cells of smokers: a pilot study. Mol Nutr Food Res 2005;49:43-51.
- Dong Z. Effects of food factors on signal transduction pathways. Biofactors 2000;12:17-28.
- Cao Y, Cao R. Angiogenesis inhibited by drinking tea. Nature 1999;398:381.
- Klaunig JE, Xu Y, Han C, Kamendulis LM, Chen J, Heiser C, et al. The effect of tea consumption on oxidative stress in smokers and nonsmokers. Proc Soc Exp Biol Med 1999;220:249-54.
- Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res 2006;66:1234-40.
- Suganuma M, Okabe S, Kai Y, Sueoka N, Sueoka E, Fujiki H. Synergistic effects of (–)-epigallocatechin gallate with (–)-epicatechin, sulindac, or tamoxifen on cancer-preventive activity in the human lung cancer cell line PC-9. Cancer Res 1999;59:44-7.
- Adhami VM, Malik A, Zaman N, Sarfaraz S, Siddiqui IA, Syed DN, et al. Combined inhibitory effects of green tea polyphenols and selective cyclooxygenase-2 inhibitors on the growth of human prostate cancer cells both in vitro and in vivo. Clin Cancer Res 2007;13:1611-9.
- Masuda M, Suzui M, Weinstein IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res 2001;7:4220-9.
- Masuda M, Suzui M, Lim JT, Weinstein IB. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clin Cancer Res 2003;9:3486-91.
- Orner GA, Dashwood WM, Blum CA, Diaz GD, Li Q, Dashwood RH. Suppression of tumorigenesis in the Apc(min) mouse: down-regulation of beta-catenin signaling by a combination of tea plus sulindac. Carcinogenesis 2003;24:263-7.
- Ohishi T, Kishimoto Y, Miura N, Shiota G, Kohri T, Hara Y, et al. Synergistic effects of (-)-epigallocatechin gallate with sulindac against colon carcinogenesis of rats treated with azoxymethane. Cancer Lett 2002;177:49-56.
- Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res 2004;24:2783-840.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006;5:493-506.
- Bhat KP, Pezzuto JM. Cancer chemopreventive activity of resveratrol. Ann N Y Acad Sci 2002;957:210-29.
- Asensi M, Medina I, Ortega A, Carretero J, Bano MC, Obrador E, et al. Inhibition of cancer growth by resveratrol is related to its low bioavailability. Free Radic Biol Med 2002;33:387-98.
- de Santi C, Pietrabissa A, Mosca F, Pacifici GM. Glucuronidation of resveratrol, a natural product present in grape and wine, in the human liver. Xenobiotica 2000;30:1047-54.
- Fremont L. Biological effects of resveratrol. Life Sci 2000;66:663-73.
- Boocock DJ, Faust GES, Patel KR, Schinas AM, Brown VA, Ducharme MP, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 2007;16:1246-52.
- Boocock DJ, Patel KR, Faust GE, Normolle DP, Marczylo TH, Crowell JA, et al. Quantitation of trans-resveratrol and detection of its metabolites in human plasma and urine by high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2007;848:182-7.
- Goldberg DM, Yan J, Soleas GJ. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clin Biochem 2003;36:79-87.
- Wang LX, Heredia A, Song H, Zhang Z, Yu B, Davis C, et al. Resveratrol glucuronides as the metabolites of resveratrol in humans: characterization, synthesis, and anti-HIV activity. J Pharm Sci 2004;93:2448-57.
- Yu C, Shin YG, Chow A, Li Y, Kosmeder JW, Lee YS, et al. Human, rat, and mouse metabolism of resveratrol. Pharm Res 2002;19:1907-14.
- Miksits M, Maier-Salamon A, Aust S, Thalhammer T, Reznicek G, Kunert O, et al. Sulfation of resveratrol in human liver: evidence of a major role for the sulfotransferases SULT1A1 and SULT1E1. Xenobiotica 2005;35:1101-19.
- Sabolovic N, Humbert AC, Radominska-Pandya A, Magdalou J. Resveratrol is efficiently glucuronidated by UDP-glucuronosyl-transferases in the human gastrointestinal tract and in Caco-2 cells. Biopharm Drug Dispos 2006;27:181-9.
- De Santi C, Pietrabissa A, Spisni R, Mosca F, Pacifici GM. Sulphation of resveratrol, a natural compound present in wine, and its inhibition by natural flavonoids. Xenobiotica 2000;30:857-66.
- Regev-Shoshani G, Shoseyov O, Bilkis I, Kerem Z. Glycosylation of resveratrol protects it from enzymic oxidation. Biochem J 2003;374:157-63.
- Kuhnle G, Spencer JP, Chowrimootoo G, Schroeter H, Debnam ES, Srai SK, et al. Resveratrol is absorbed in the small intestine as resveratrol glucuronide. Biochem Biophys Res Commun 2000;272:212-7.
- Jannin B, Menzel M, Berlot JP, Delmas D, Lancon A, Latruffe N. Transport of resveratrol, a cancer chemopreventive agent, to cellular targets: plasmatic protein binding and cell uptake. Biochem Pharmacol 2004;68:1113-8.
- Marier JF, Vachon P, Gritsas A, Zhang J, Moreau JP, Ducharme MP. Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J Pharmacol Exp Ther 2002;302:369-73.
- Vitrac X, Desmouliere A, Brouillaud B, Krisa S, Deffieux G, Barthe N, et al. Distribution of [14C]-trans-resveratrol, a cancer chemo-preventive polyphenol, in mouse tissues after oral administration. Life Sci 2003;72:2219-33.
- Sale S, Verschoyle RD, Boocock D, Jones DJ, Wilsher N, Ruparelia KC, et al. Pharmacokinetics in mice and growth-inhibitory properties of the putative cancer chemopreventive agent resvera-trol and the synthetic analogue trans 3,4,5,4'-tetramethoxy-stilbene. Br J Cancer 2004;90:736-44.
- Abd El-Mohsen M, Bayele H, Kuhnle G, Gibson G, Debnam E, Kaila Srai S, et al. Distribution of [3H]trans-resveratrol in rat tissues following oral administration. Brit J Nutr 2006;96:62-70.
- Wenzel E, Soldo T, Erbersdobler H, Somoza V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol Nutr Food Res 2005;49:482-94.
- Poolman TM, Ng LL, Farmer PB, Manson MM. Inhibition of the respiratory burst by resveratrol in human monocytes: correlation with inhibition of PI3K signaling. Free Radic Biol Med 2005;39:118-32.
- Whitehead TP, Robinson D, Allaway S, Syms J, Hale A. Effect of red wine ingestion on the antioxidant capacity of serum. Clin Chem 1995;41:32-5.
- Garvin S, Ollinger K, Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett 2006;231:113-22.
- Carbo N, Costelli P, Baccino FM, Lopez-Soriano FJ, Argiles JM. Resveratrol, a natural product present in wine, decreases tumour growth in a rat tumour model. Biochem Biophys Res Commun 1999;254:739-43.
- Chen Y, Tseng SH, Lai HS, Chen WJ. Resveratrol-induced cellular apoptosis and cell cycle arrest in neuroblastoma cells and antitumor effects on neuroblastoma in mice. Surgery 2004;136:57-66.
- Tseng SH, Lin SM, Chen JC, Su YH, Huang HY, Chen CK, et al. Resveratrol suppresses the angiogenesis and tumor growth of gliomas in rats. Clin Cancer Res 2004;10:2190-202.
- Banerjee S, Bueso-Ramos C, Aggarwal BB. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: role of nuclear factor-kappaB, cyclooxy-genase 2, and matrix metalloprotease 9. Cancer Res 2002;62:4945-54.
- Martin AR, Villegas I, Sanchez-Hidalgo M, de la Lastra CA. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br J Pharmacol 2006;147:873-85.
- Sale S, Tunstall RG, Ruparelia KC, Potter GA, Steward WP, Gescher AJ. Comparison of the effects of the chemopreventive agent resveratrol and its synthetic analog trans 3,4,5,4'-tetra-methoxystilbene (DMU-212) on adenoma development in the Apc(Min+) mouse and cyclooxygenase-2 in human-derived colon cancer cells. Int J Cancer 2005;115:194-201.
- Provinciali M, Re F, Donnini A, Orlando F, Bartozzi B, Di Stasio G, et al. Effect of resveratrol on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Int J Cancer 2005;115:36-45.
- Tessitore L, Davit A, Sarotto I, Caderni G. Resveratrol depresses the growth of colorectal aberrant crypt foci by affecting bax and p21(CIP) expression. Carcinogenesis 2000;21:1619-22.
- Sengottuvelan M, Viswanathan P, Nalini N. Chemopreventive effect of trans-resveratrol — a phytoalexin against colonic aberrant crypt foci and cell proliferation in 1,2-dimethylhydrazine induced colon carcinogenesis. Carcinogenesis 2006;27:1038-46.
- Kweon S, Kim Y, Choi H. Grape extracts suppress the formation of preneoplastic foci and activity of fatty acid synthase in rat liver. Exp Mol Med 2003;35:371-8.
- Delmas D, Rebe C, Micheau O, Athias A, Gambert P, Grazide S, et al. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene 2004;23:8979-86.
- Fulda S, Debatin KM. Sensitization for anticancer drug-induced apoptosis by the chemopreventive agent resveratrol. Oncogene 2004;23:6702-11.
- Fulda S, Debatin KM. Resveratrol-mediated sensitisation to TRAIL-induced apoptosis depends on death receptor and mitochondrial signalling. Eur J Cancer 2005;41:786-98.
- Kubota T, Uemura Y, Kobayashi M, Taguchi H. Combined effects of resveratrol and paclitaxel on lung cancer cells. Anticancer Res 2003;23:4039-46.
- Rezk YA, Balulad SS, Keller RS, Bennett JA. Use of resveratrol to improve the effectiveness of cisplatin and doxorubicin: study in human gynecologic cancer cell lines and in rodent heart. Am J Obstet Gynecol 2006;194:e23-6.
- Awad AB, Burr AT, Fink CS. Effect of resveratrol and beta-sitosterol in combination on reactive oxygen species and prostaglandin release by PC-3 cells. Prostag Leukotr Ess 2005;72:219-26.
- Mertens-Talcott SU, Percival SS. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett 2005;218:141-51.
- Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3-carbinol and 3,3' diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem Biophys Res Comm 2005;328:63-9.
- Howells LM, Gallacher-Horley B, Houghton CE, Manson MM, Hudson EA. Indole-3-carbinol inhibits protein kinase B/Akt and induces apoptosis in the human breast tumor cell line MDA MB468 but not in the nontumorigenic HBL100 line. Mol Cancer Ther 2002;1:1161-72.
- Lee IJ, Han F, Baek J, Hisatsune A, Kim KC. Inhibition of MUC1 expression by indole-3-carbinol. Int J Cancer 2004;109:810-16.
- Telang NT, Katdare M, Bradlow HL, Osborne MP, Fishman J. Inhibition of proliferation and modulation of estradiol metabolism: novel mechanisms for breast cancer prevention by the phytochemical indole-3-carbinol. Proc Soc Exp Biol Med 1997;216:246-52.
- Rahman KMW, Aranha O, Glazyrin A, Chinni SR, Sarkar FH. Translocation of Bax to mitochondria induces apoptotic cell death in Indole-3-carbinol (I3C) treated breast cancer cells. Oncogene 2000;19:5764-71.
- Ge X, Fares FA, Yannai S. Induction of apoptosis in MCF-7 cells by indole-3-carbinol is independent of p53 and Bax. Anticancer Res 1999;19:3199-204.
- Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, et al. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signalling. J Biol Chem 1998;273:3838-47.
- Ashok BT, Chen Y, Liu X, Bradlow HL, Mittelman A, Tiwari RK. Abrogation of estrogen-mediated cellular and biochemical effects by indole-3-carbinol. Nutr Cancer 2001;41:180-7.
- Niwa T, Swaneck G, Bradlow HL. Alterations in estradiol metabolism in MCF-7 cells induced by treatment with indole-3-carbinol and related compounds. Steroids 1994;59:523-7.
- Tiwari RK, Guo L, Bradlow HL, Telang NT, Osborne MP. Selective responsiveness of human breast cancer cells to indole-3-carbinol, a chemopreventive agent. J Natl Cancer Inst 1994;86:126-31.
- Ashok BT, Chen YG, Liu X, Garikapaty VPS, Seplowitz R, Tschorn J, et al. Multiple molecular targets of indole-3-carbinol, a chemopreventive anti-estrogen in breast cancer. Eur J Cancer Prev 2002;11:S86-93.
- Garcia HH, Brar GA, Nguyen DHH, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribu-tion, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem 2005;280:8756-64.
- Rahman KMW, Aranha OP, Sarkar FH. Indole-3-carbinol (I3C) induces apoptosis in tumorigenic but not in nontumorigenic breast epithelial cells. Nutr Cancer 2003;45:101-12.
- Meng QH, Goldberg ID, Rosen EM, Fan SJ. Inhibitory effects of indole-3-carbinol on invasion and migration in human breast cancer cells. Breast Cancer Res Treat 2000;63:147-52.
- Chinni SR, Sarkar FH. Akt inactivation is a key event in indole-3-carbinol-induced apoptosis in PC-3 cells. Clin Cancer Res 2002;8:1228-36.
- Jeon KI, Rih JK, Kim HJ, Lee YJ, Cho CH, Goldberg ID, et al. Pretreatment of indole-3-carbinol augments TRAIL-induced apoptosis in a prostate cancer cell line, LNCaP. FEBS Lett 2003;544:246-51.
- Pappa G, Lichtenberg M, Iori R, Barillari J, Bartsch H, Gerhauser C. Comparison of growth inhibition profiles and mechanisms of apoptosis induction in human colon cancer cell lines by isothiocyanates and indoles from Brassicaceae. Mutat Res 2006;599:76-87.
- Hong C, Firestone GL, Bjeldanes LF. Bcl-2 family-mediated apoptotic effects of 3,3'-diindolylmethane (DIM) in human breast cancer cells. Biochem Pharmacol 2002;63:1085-97.
- Ge X, Yannai S, Rennert G, Gruener N, Fares FA. 3,3'-Diindolyl-methane induces apoptosis in human cancer cells. Biochem Biophys Res Commun 1996;228:153-8.
- Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3'-Diindolyl-methane (DIM) induces a G(1) cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis 2002;23:1297-305.
- Rahman KW, Li Y, Wang Z, Sarkar SH, Sarkar FH. Gene expres-sion profiling revealed survivin as a target of 3,3'-diindolyl-methane-induced cell growth inhibition and apoptosis in breast cancer cells. Cancer Res 2006;66:4952-60.
- Nachshon-Kedmi M, Yannai S, Haj A, Fares FA. Indole-3-carbinol and 3,3'-diindolylmethane induce apoptosis in human prostate cancer cells. Food Chem Toxicol 2003;41:745-52.
- Garikapaty VP, Ashok BT, Tadi K, Mittelman A, Tiwari RK. 3,3'-Diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem Biophys Res Commun 2006;340:718-25.
- Gamet-Payrastre L, Lumeau S, Gasc N, Cassar G, Rollin P, Tulliez J. Selective cytostatic and cytotoxic effects of glucosinolates hydrolysis products on human colon cancer cells in vitro. Anti-cancer Drugs 1998;9:141-8.
- Sanderson JT, Slobbe L, Lansbergen GWA, Safe S, van den Berg M. 2,3,7,8-tetrachlorodibenzo-p-dioxin and diindolylmethanes differentially induce cytochrome P450 1A1, 1B1, and 19 in H295R human adrenocortical carcinoma cells. Toxicol Sci 2001;61:40-8.
- Garcea G, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, et al. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br J Cancer 2004;90:1011-5.
- Lao CD. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med 2006;6:10.
- Wahlstrom B, Blennow G. A study on the fate of curcumin in the rat. Acta Pharmacol Toxicol (Copenh) 1978;43:86-92.
- Holder GM, Plummer JL, Ryan AJ. The metabolism and excretion of curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) in the rat. Xenobiotica 1978;8:761-8.
- Ravindranath V, Chandrasekhara N. Metabolism of curcumin—studies with [3H]curcumin. Toxicol 1981;22:337-44.
- Pan MH, Huang TM, Lin JK. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab Dispos 1999;27:486-94.
- Yang KY, Lin LC, Tseng TY, Wang SC, Tsai TH. Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 2007;853:183-9.
- Mori H, Niwa K, Zheng Q, Yamada Y, Sakata K, Yoshimi N. Cell proliferation in cancer prevention; effects of preventive agents on estrogen-related endometrial carcinogenesis model and on an in vitro model in human colorectal cells. Mutat Res 2001;480–481:201-7.
- Collett GP, Campbell FC. Curcumin induces c-jun N-terminal kinase-dependent apoptosis in HCT116 human colon cancer cells. Carcinogenesis 2004;25:2183-9.
- Scott DW, Loo G. Curcumin-induced GADD153 gene up-regulation in human colon cancer cells. Carcinogenesis 2004;25:2155-64.
- Chen A, Xu J. Activation of PPAR{gamma} by curcumin inhibits Moser cell growth and mediates suppression of gene expression of cyclin D1 and EGFR. Am J Physiol Gastrointest Liver Physiol 2005;288:G447-56.
- Chen A, Xu J, Johnson AC. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006;25:278-87.
- Zhang F, Altorki NK, Mestre JR, Subbaramaiah K, Dannenberg AJ. Curcumin inhibits cyclooxygenase-2 transcription in bile acid- and phorbol ester-treated human gastrointestinal epithelial cells. Carcinogenesis 1999;20:445-51.
- Plummer SM, Holloway KA, Manson MM, Munks RJ, Kaptein A, Farrow S, et al. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene 1999;18:6013-20.
- Goel A, Boland CR, Chauhan DP. Specific inhibition of cyclooxygenase-2 (COX-2) expression by dietary curcumin in HT-29 human colon cancer cells. Cancer Lett 2001;172:111-8.
- Hong J, Bose M, Ju J, Ryu JH, Chen X, Sang S, et al. Modulation of arachidonic acid metabolism by curcumin and related beta-diketone derivatives: effects on cytosolic phospholipase A(2), cyclooxygenases and 5-lipoxygenase. Carcinogenesis 2004;25:1671-9.
- Lin LI, Ke YF, Ko YC, Lin JK. Curcumin inhibits SK-Hep-1 hepatocellular carcinoma cell invasion in vitro and suppresses matrix metalloproteinase-9 secretion. Oncol 1998;55:349-53.
- Choudhuri T, Pal S, Agwarwal ML, Das T, Sa G. Curcumin induces apoptosis in human breast cancer cells through p53-dependent Bax induction. FEBS Lett 2002;512:334-40.
- Simon A, Allais DP, Duroux JL, Basly JP, Durand-Fontanier S, Delage C. Inhibitory effect of curcuminoids on MCF-7 cell proliferation and structure-activity relationships. Cancer Lett 1998;129:111-6.
- Anuchapreeda S, Leechanachai P, Smith MM, Ambudkar SV, Limtrakul PN. Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem Pharmacol 2002;64:573-82.
- Han SS, Chung ST, Robertson DA, Ranjan D, Bondada S. Curcumin causes the growth arrest and apoptosis of B cell lymphoma by downregulation of egr-1, c-myc, bcl-XL, NF-kappa B, and p53. Clin Immunol 1999;93:152-61.
- Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998;17:173-8.
- Kuo ML, Huang TS, Lin JK. Curcumin, an antioxidant and anti-tumor promoter, induces apoptosis in human leukemia cells. Biochim Biophys Acta 1996;1317:95-100.
- Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloyl-methane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol 2003;171:3863-71.
- Yang CS, Chen L, Lee MJ, Balentine D, Kuo MC, Schantz SP. Blood and urine levels of tea catechins after ingestion of different amounts of green tea by human volunteers. Cancer Epidemiol Biomarkers Prev 1998;7:351-4.
- Meng X, Sang S, Zhu N, Lu H, Sheng S, Lee MJ, et al. Identification and characterization of methylated and ring-fission metabolites of tea catechins formed in humans, mice, and rats. Chem Res Toxicol 2002;15:1042-50.
- Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, et al. Pharmacokinetics of tea catechins after ingestion of green tea and (-)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev 2002;11:1025-32.
- Ullmann U, Haller J, Decourt JP, Girault N, Girault J, Richard-Caudron AS, et al. A single ascending dose study of epigallo-catechin gallate in healthy volunteers. J Int Med Res 2003;31:88-101.
- Van Amelsvoort JM, Van Hof KH, Mathot JN, Mulder TP, Wiersma A, Tijburg LB. Plasma concentrations of individual tea catechins after a single oral dose in humans. Xenobiotica 2001;31:891-901.
- Warden BA, Smith LS, Beecher GR, Balentine DA, Clevidence BA. Catechins are bioavailable in men and women drinking black tea throughout the day. J Nutr 2001;131:1731-7.
- Chow HH, Hakim IA, Vining DR, Crowell JA, Ranger-Moore J, Chew WM, et al. Effects of dosing condition on the oral bioavailability of green tea catechins after single-dose administration of Polyphenon E in healthy individuals. Clin Cancer Res 2005;11:4627-33.
- Henning SM, Niu Y, Liu Y, Lee NH, Hara Y, Thames GD, et al. Bioavailability and antioxidant effect of epigallocatechin gallate administered in purified form versus as green tea extract in healthy individuals. J Nutr Biochem 2005;16:610-6.
- Lambert JD, Lee MJ, Lu H, Meng X, Hong JJ, Seril DN, et al. Epigallocatechin-3-gallate is absorbed but extensively glucuro-nidated following oral administration to mice. J Nutr 2003;133:4172-7.
- Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, elimination of tea polyphenols in rats. Drug Metab Dispos 1997;25:1045-50.
- Jeong WS, Kim IW, Hu R, Kong AN. Modulation of AP-1 by natural chemopreventive compounds in human colon HT-29 cancer cell line. Pharm Res 2004;21:649-60.
- Jung YD, Kim MS, Shin BA, Chay KO, Ahn BW, Liu W, et al. EGCG, a major component of green tea, inhibits tumour growth by inhibiting VEGF induction in human colon carcinoma cells. Br J Cancer 2001;84:844-50.
- McLoughlin P, Roengvoraphoj M, Gissel C, Hescheler J, Certa U, Sachinidis A. Transcriptional responses to epigallocatechin-3-gallate in HT 29 colon carcinoma spheroids. Genes Cells 2004;9:661-9.
- Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 2001;276:13322-30.
- Hussain T, Gupta S, Adhami VM, Mukhtar H. Green tea constituent epigallocatechin-3-gallate selectively inhibits COX-2 without affecting COX-1 expression in human prostate carcinoma cells. Int J Cancer 2005;113:660-9.
- Siddiqui IA, Adhami VM, Afaq F, Ahmad N, Mukhtar H. Modulation of phosphatidylinositol-3-kinase/protein kinase B- and mitogen-activated protein kinase-pathways by tea polyphenols in human prostate cancer cells. J Cell Biochem 2004;91:232-42.
- Gupta S, Ahmad N, Nieminen AL, Mukhtar H. Growth inhibition, cell-cycle dysregulation, and induction of apoptosis by green tea constituent (-)-epigallocatechin-3-gallate in androgen-sensitive and androgen-insensitive human prostate carcinoma cells. Toxicol Appl Pharmacol 2000;164:82-90.
- Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood 2004;104:788-94.
- Balasubramanian S, Efimova T, Eckert RL. Green tea polyphenol stimulates a Ras, MEKK1, MEK3, and p38 cascade to increase activator protein 1 factor-dependent involucrin gene expression in normal human keratinocytes. J Biol Chem 2002;277:1828-36.
- Barthelman M, Bair WB 3rd, Stickland KK, Chen W, Timmermann BN, Valcic S, et al. (-)-Epigallocatechin-3-gallate inhibition of ultraviolet B-induced AP-1 activity. Carcinogenesis 1998;19:2201-4.
- Chen W, Dong Z, Valcic S, Timmermann BN, Bowden GT. Inhibition of ultraviolet B — induced c-fos gene expression and p38 mitogen-activated protein kinase activation by (-)-epigallo-catechin gallate in a human keratinocyte cell line. Mol Carcinog 1999;24:79-84.
- Nihal M, Ahmad N, Mukhtar H, Wood GS. Anti-proliferative and proapoptotic effects of (-)-epigallocatechin-3-gallate on human melanoma: possible implications for the chemoprevention of melanoma. Int J Cancer 2005;114:513-21.
- Nomura M, Kaji A, He Z, Ma WY, Miyamoto K, Yang CS, et al. Inhibitory mechanisms of tea polyphenols on the ultraviolet B-activated phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 2001;276:46624-31.
- Nomura M, Ma W, Chen N, Bode AM, Dong Z. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced NF-kappaB activation by tea polyphenols, (-)-epigallocatechin gallate and theaflavins. Carcinogenesis 2000;21:1885-90.
- Dong Z, Ma W, Huang C, Yang CS. Inhibition of tumor promoter-induced activator protein 1 activation and cell transformation by tea polyphenols, (-)-epigallocatechin gallate, and theaflavins. Cancer Res 1997;57:4414-9.
- Liang YC, Lin-shiau SY, Chen CF, Lin JK. Suppression of extracellular signals and cell proliferation through EGF receptor binding by (-)-epigallocatechin gallate in human A431 epidermoid carcinoma cells. J Cell Biochem 1997;67:55-65.
- Gupta S, Hastak K, Afaq F, Ahmad N, Mukhtar H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene 2004;23:2507-22.
- Ahmad N, Gupta S, Mukhtar H. Green tea polyphenol epigallo-catechin-3-gallate differentially modulates nuclear factor kappaB in cancer cells versus normal cells. Arch Biochem Biophys 2000;376:338-46.
- Sah JF, Balasubramanian S, Eckert RL, Rorke EA. Epigallo-catechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J Biol Chem 2004;279:12755-62.
- Bhimani RS, Troll W, Grunberger D, Frenkel K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res 1993;53:4528-33.
- Chen JJ, Ye ZQ, Koo MW. Growth inhibition and cell cycle arrest effects of epigallocatechin gallate in the NBT-II bladder tumour cell line. BJU Int 2004;93:1082-6.
- Khafif A, Schantz SP, al-Rawi M, Edelstein D, Sacks PG. Green tea regulates cell cycle progression in oral leukoplakia. Head Neck 1998;20:528-34.
- Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea poly-phenol (-)-epigallocatechin-3-gallate inhibits DNA methyltrans-ferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003;63:7563-70.
- Lamy S, Gingras D, Beliveau R. Green tea catechins inhibit vascular endothelial growth factor receptor phosphorylation. Cancer Res 2002;62:381-5.
- Tang FY, Nguyen N, Meydani M. Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. Int J Cancer 2003;106:871-8.
- El Bedoui J, Oak MH, Anglard P, Schini-Kerth VB. Catechins prevent vascular smooth muscle cell invasion by inhibiting MT1–MMP activity and MMP-2 expression. Cardiovasc Res 2005;67:317-25.
- Dashwood WM, Carter O, Al-Fageeh M, Li Q, Dashwood RH. Lysosomal trafficking of beta-catenin induced by the tea polyphenol epigallocatechin-3-gallate. Mutat Res 2005;591:161-72.
- Moon Y, Lee M, Yang H. Involvement of early growth response gene 1 in the modulation of microsomal prostaglandin E synthase 1 by epigallocatechin gallate in A549 human pulmonary epithelial cells. Biochem Pharmacol 2007;73:125-35.
- Yu HN, Shen SR, Yin JJ. Effects of interactions of EGCG and Cd(2+) on the growth of PC-3 cells and their mechanisms. Food Chem Toxicol 2007;45:244-9.
- Kalfon L, Youdim MB, Mandel SA. Green tea polyphenol (–)-epigallocatechin-3-gallate promotes the rapid protein kinase C- and proteasome-mediated degradation of Bad: implications for neuroprotection. J Neurochem 2007;100:992-1002.
- Walle T, Hsieh F, DeLegge MH, Oatis JE Jr, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos 2004;32:1377-82.
- Vitaglione P, Sforza S, Galaverna G, Ghidini C, Caporaso N, Vescovi PP, et al. Bioavailability of trans-resveratrol from red wine in humans. Mol Nutr Food Res 2005;49:495-504.
- Meng X, Maliakal P, Lu H, Lee MJ, Yang CS. Urinary and plasma levels of resveratrol and quercetin in humans, mice, and rats after ingestion of pure compounds and grape juice. J Agric Food Chem 2004;52:935-42.
- Juan ME, Lamuela-Raventos RM, de la Torre-Boronat MC, Planas JM. Determination of trans-resveratrol in plasma by HPLC. Anal Chem 1999;71:747-50.
- Hsieh TC, Wu JM. Differential effects on growth, cell cycle arrest, and induction of apoptosis by resveratrol in human prostate cancer cell lines. Exp Cell Res 1999;249:109-15.
- Aziz MH, Kumar R, Ahmad N. Cancer chemoprevention by resveratrol: In vitro and in vivo studies and the underlying mechanisms. Int J Oncol 2003;23:17-28.
- Cao Z, Fang J, Xia C, Shi X, Jiang BH. -3,4,5'-Trihydroxy-stibene inhibits hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in human ovarian cancer cells. Clin Cancer Res 2004;10:5253-63.
- Kaneuchi M, Sasaki M, Tanaka Y, Yamamoto R, Sakuragi N, Dahiya R. Resveratrol suppresses growth of Ishikawa cells through down-regulation of EGF. Int J Oncol 2003;23:1167-72.
- Lee JE, Safe S. Involvement of a post-transcriptional mechanism in the inhibition of CYP1A1 expression by resveratrol in breast cancer cells. Biochem Pharmacol 2001;62:1113-24.
- El-Mowafy AM, Alkhalaf M. Resveratrol activates adenylyl-cyclase in human breast cancer cells: a novel, estrogen receptor-independent cytostatic mechanism. Carcinogenesis 2003;24:869-73.
- Bhat KP, Lantvit D, Christov K, Mehta RG, Moon RC, Pezzuto JM. Estrogenic aps-28-09-1499and antiestrogenic properties of resveratrol in mammary tumor models. Cancer Res 2001;61:7456-63.
- Basly JP, Marre-Fournier F, Le Bail JC, Habrioux G, Chulia AJ. Estrogenic/antiestrogenic and scavenging properties of (E)- and (Z)-resveratrol. Life Sci 2000;66:769-77.
- Scarlatti F, Sala G, Somenzi G, Signorelli P, Sacchi N, Ghidoni R. Resveratrol induces growth inhibition and apoptosis in metastatic breast cancer cells via de novo ceramide signaling. FASEB J 2003;17:2339-41.
- Subbaramaiah K, Chung WJ, Michaluart P, Telang N, Tanabe T, Inoue H, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem 1998;273:21875-82.
- Shih A, Davis FB, Lin HY, Davis PJ. Resveratrol induces apoptosis in thyroid cancer cell lines via a MAPK- and p53-dependent mechanism. J Clin Endocrinol Metab 2002;87:1223-32.
- She QB, Huang C, Zhang Y, Dong Z. Involvement of c-jun NH(2)-terminal kinases in resveratrol-induced activation of p53 and apoptosis. Mol Carcinogenesis 2002;33:244-50.
- She QB, Ma WY, Wang M, Kaji A, Ho CT, Dong Z. Inhibition of cell transformation by resveratrol and its derivatives: differential effects and mechanisms involved. Oncogene 2003;22:2143-50.
- Woo JH, Lim JH, Kim YH, Suh SI, Min DS, Chang JS, et al. Resveratrol inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting JNK and PKC delta signal transduction. Oncogene 2004;23:1845-53.
- Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett 2001;171:11-6.
- Ahmad N, Adhami VM, Afaq F, Feyes DK, Mukhtar H. Resveratrol causes WAF-1/p21-mediated G(1)-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin Cancer Res 2001;7:1466-73.
- Ahmad KA, Clement MV, Hanif IM, Pervaiz S. Resveratrol inhibits drug-induced apoptosis in human leukemia cells by creating an intracellular milieu nonpermissive for death execution. Cancer Res 2004;64:1452-59.
- Huang C, Ma WY, Goranson A, Dong Z. Resveratrol suppresses cell transformation and induces apoptosis through a p53-dependent pathway. Carcinogenesis 1999;20:237-42.
- Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol 2000;164:6509-19.
- Berge G, Ovrebo S, Botnen IV, Hewer A, Phillips DH, Haugen A, et al. Resveratrol inhibits benzo[a]pyrene-DNA adduct formation in human bronchial epithelial cells. Br J Cancer 2004;91:333-8.
- Hsieh TC, Wang Z, Hamby CV, Wu JM. Inhibition of melanoma cell proliferation by resveratrol is correlated with upregula-tion of quinone reductase 2 and p53. Biochem Biophys Res Commun 2005;334:223-30.
- Zhang Q, Tang X, Lu QY, Zhang ZF, Brown J, Le AD. Resvera-trol inhibits hypoxia-induced accumulation of hypoxia-inducible factor-1alpha and VEGF expression in human tongue squamous cell carcinoma and hepatoma cells. Mol Cancer Ther 2005;4:1465-74.