
Biomarkers for diet and cancer prevention research: potentials and challenges
Introduction
Dietary habits are recognized to be an important modifiable environmental factor influencing cancer risk and tumor behavior. Although some researchers have estimated that about 30%–40% of all cancer cases relate to dietary habits, the actual percentage is highly dependent on the foods consumed and the specific type of cancer[1]. Both essential and non-essential allelochemicals arising from plants, along with zoochemicals occurring in animal products, fungochemicals from mushrooms, and bacterochemicals from bacteria may be physiologically relevant modifiers of cancer risk. Compounds encompassing such diverse categories as minerals, amino acids, carbohydrates, fatty acids, carotenoids, dithiolthiones, flavonoids, terpenes, glucosinolates, isothio-cyanates and allyl sulfurs may influence multiple pathways involved with cancer, many of which may act additively or synergistically when combined in the human diet.
While optimizing the intake of specific foods and/or their bioactive components seems a prudent, non-invasive and cost-effective strategy for reducing the cancer burden, this is far from a simple process[2]. The magnitude of the problem in identifying critical dietary components is evident by the literally thousands of compounds consumed each day[2,3]. Furthermore, the dearth of quantitative information about some food constituents limits the ability to unravel which constituents are most important. Although it is estimated that humans consume >5000 individual flavonoids, only a few have been examined for their cancer protective effects[2]. Unfortunately many food phytochemicals remain largely uncharacterized and this can lead to confusion about the true role of diet in determining health. Interactions between the different components within a food may explain why isolated components do not always result in similar biological outcomes to the intact food[4]. Likewise, interactions among foods and their constituents may contribute to the overall relationship between eating behaviors and cancer[4].
Scientifically sound intervention studies must be viewed as the cornerstone for establishing nutrition guidance[5]. Unfortunately, the number of long-term intervention studies that would be needed to adequately define the needs for bioactive food components is likely to be impractical in terms of speed of discovery and cost. Alternative procedures that use validated and sensitive biological markers are needed that can serve as predictors of those who will benefit most and those who might be placed at risk, and the minimum quantity of the food and/or component required to bring about the intended response. Undeniably, the identification and selection of appropriate biomarkers will not be an easy task, but if successful it will have enormous societal importance.
A biomarker can potentially be any substance, structure or process that could be monitored in tissues or fluids and that predicts or influences health, or assesses the incidence or biological behavior of a disease[5]. Identification of biomarkers that are on the causal pathway, have a high probability of reflecting health or the progression to clinical disease, and have the ability to account for all or most of the variation in a physiological state or the preponderance of cases of the specified clinical outcome, have largely remained elusive[5]. Classes of biomarkers required to adequately evaluate the physiological significance of functional foods are probably no different than those used to evaluate environmental factors that might influence health or disease risk[5–8]. Thus, biomarkers are needed that reliably evaluate “intake” or exposure to a specific food or its component, assess one or more specific biological “effects”, and effectively predict individual “susceptibility” (Figure 1). The interdependence of these three categories of biomarkers cannot be over-emphasized.
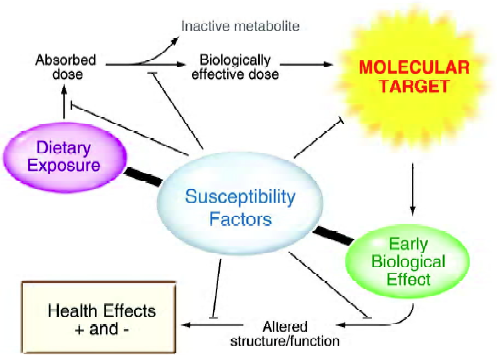
The selection and validation of intake/exposure, effect and susceptibility biomarkers must build on sound scientific investigations and ideally they should be accepted universally for their predictability. These biomarkers must be readily accessible, easily and reliably assayed, differentially expressed in normal and malignant tissues, directly associated with disease progression, modifiable and – most importantly – predictive. The utility of biomarkers capable of predicting cancer risk will be most valuable if they confer a long lead-time relative to the onset of the disease.
Molecular biomarkers (the “-omics” approach) will likely offer the sensitivity and reliability to evaluate dietary exposures and to provide invaluable insights into behaviors of specific molecular targets and predictors of individual responsiveness to dietary change[2]. The study of nutri-genomics has the potential to identify definitively which components in foods bring about either positive or negative consequences, and to clarify their relevant mechanisms of action and most importantly when they can be manipulated to reduce cancer risk[9–11]. Knowledge about how diet-induced phenotypic responses depend on an individual’s genetic background (nutrigenetics), the expression of genes (epigenomics and transcriptomics), changes in the amounts and activities of proteins (proteomics), and shifts in small molecular weight compounds (metabolomics) – collectively referred to as “-omics” – will be important in identifying responders from non-responders[9–11].
Intake or exposure biomarkers
Rapid, accurate and inexpensive methods for assessing the dietary intake of specific nutrients, both essential and non-essential, remain a cornerstone, and yet present a challenge, to determining the relationship between dietary intake and cancer risk. Errors in estimating food intakes, interactions among food components and incomplete data on the content of nutrients limit the usefulness of self-reported data of food consumption as an “exposure” biomarker. Because diet is exceedingly complex and may consist of foods that are consumed intermittently and irregularly, self-reports of diet may be particularly prone to measurement error. Food frequency questionnaires (FFQ) and 24-h recalls are two of the major dietary data collection instruments that are often used to estimate exposure.
The FFQ is the most commonly used diet assessment instrument in large observational studies. FFQ are designed to measure a person’s usual dietary intake over a defined period of time. FFQ are convenient, measure long-term behavior and are relatively inexpensive. However, FFQ are limited by knowledge about particular foods and are hampered by the inability of individuals to accurately report their food intake retrospectively over a long period of time[12]. FFQ are prone to underreporting of energy and protein by 20%–50%[13]. Analytical issues compound this problem when microconstituents are being considered for their impact on health.
In contrast, 24-h recalls provide in-depth information about the type and amount of foods consumed; however, intake on a single day is a poor estimator of long-term usual intake[12]. Over 20 years ago, Guthrie and Crocetti[14] found considerable variability in daily intakes of essential nutrients among individuals over a 3-d period. In their study, less than 50% of individuals had daily intakes that were within ±25% for most nutrients. Although multiple 24-h recalls or food diaries overcome these limitations, cost can become prohibitive in terms of expense, time commitment and personnel involvement[12,15]. Unfortunately older assessment tools failed to adequately evaluate dietary supplements as a contributor to the intake of essential and non-essential nutrients. Although new assessment instruments and novel statistical analyses are being developed in an attempt to overcome these limitations their accuracy and precision remain uncertain. The Food Propensity Questionnaire has been developed and uses frequency data as a covariate in supplementing multiple recalls for estimating the usual intake of food groups[16]. Conceptually, it may be possible to model the relationship between 24-h recall probabilities and FFQ frequencies at an aggregate level and then develop probabilities of consumption for the individual. Unfortunately, a clear relationship does not appear to hold for all foods. Finally, the degree of precision that is added by the use of the Food Propensity Questionnaire appears somewhat nominal and, therefore, its utility remains uncertain.
The validity of dietary assessment methods can be estimated by comparing measurements obtained with different estimates to biochemical markers reflecting dietary intake. In the European Prospective Investigation into Cancer (EPIC) study, correlations between 2 validated biomarkers of intake and 24-h urinary nitrogen and potassium excretion correlated better with dietary intake assessed with the 7-d food diary (r=0.57–0.67 and r=0.51–0.55, respectively) compared to those from the FFQ (r=0.21–0.29 and r=0.32–0.34, respectively)[17]. These data indicate that despite the increased subject burden and cost, the 7-d diary provided a better estimate of both protein and potassium intakes than did the FFQ. However, the low “r values” reflect a lack of sensitivity, even with the 7-d food diaries, and suggest that better biomarkers for assessing intake/exposure are needed.
It is certainly reasonable to assume that the intakes of phytochemicals and other minor dietary constituents will be even more variable when exposure is estimated based on self-reported data. Analytical problems associated with the compositional analysis of foods are in no way a trivial issue. Furthermore, some dietary components depend extensively on where the animal was raised or where the plant was grown. For example, the selenium content of food varies depending on the selenium content of the soil[18]. Plant genotype may also modify the concentration of phytochemicals. Glucora-phanin concentrations in broccoli vary more than 25-fold depending on the broccoli genotype[19].
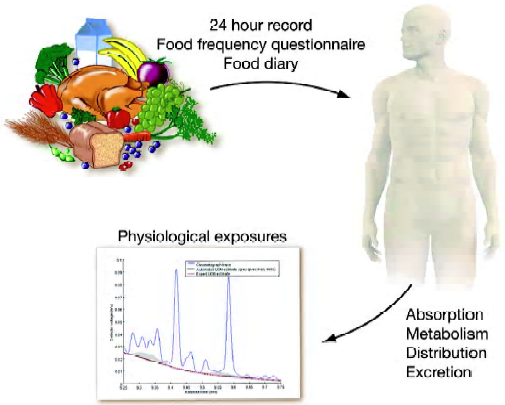
Evidence is also emerging that phytochemical absorption is highly dependent on the food source[20], as well as the method of processing, as observed with the increased lycopene availability from processed tomatoes, but the decreased cancer-protective properties of heated garlic[21,22]. It is essential that additional attention is devoted not only to the quantification of all dietary components, but also to enhancing our understanding about how home or commercial processing can influence their utilization. Absorption, metabolism, distribution and excretion may all contribute to the actual exposure of the target tissue to the bioactive component (Figure 2). Combining intake assessments with tissue or fluid concentrations may offer special insights into how genetics and other environmental factors can influence absorption and metabolism and can, thus, be useful in qualitatively approximating an individual’s exposure. Further-more, the reliability of an exposure biomarker will surely depend on a host of factors, including the time of sampling relative to when the compound was consumed, the pattern of intake and the pharmacokinetic properties of the compound being analyzed.
Effect biomarkers
An “effect” biomarker indicates the presence and magnitude of a biological response to an intake or exposure. In the case of foods/bioactive components it would provide an assessment of the impact of one or more active compounds on a physiological process and indicate a possible beneficial or adverse health effect.
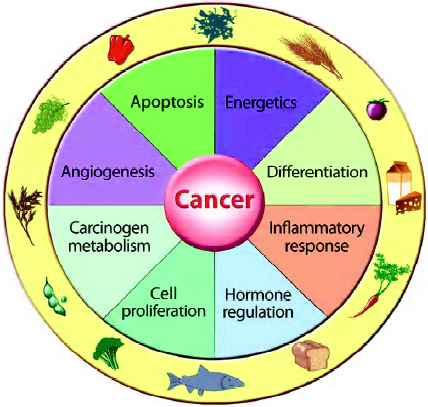
Multiple cellular processes appear to account for the response to bioactive food components in foods or dietary supplements for influencing cancer growth and/or tumor behavior (Figure 3). These include, but are not limited to, carcinogen metabolism, DNA repair, cell proliferation, programmed cell death, inflammation, differentiation and angiogenesis. As multiple responses may occur simultaneously, it is difficult to determine which is most important in dictating the overall biological response. Further-more, the ability of several nutrients to influence the same or multiple biological processes raises issues about possible synergy – as well as antagonistic interactions – that may occur within and among foods[4].
DNA instability Human cancers exhibit genomic instability and an increased mutation rate because of underlying defects in DNA repair genes. DNA mismatch repair (MMR) genes have been found to be involved in promoting cytotoxicity, apoptosis, p53 phosphorylation and cell cycle arrest following exposure to exogenous DNA damaging agents. Loss of MMR function prevents the correction of replicative errors, leading to instability of the genome, and can be detected by polymorphisms in microsatellites. A host of factors can contribute to DNA instability. Endogenous agents, including methylating species and reactive oxygen species arising during normal cellular respiration, can lead to DNA damage. Some nutrients, such as unsaturated fatty acids and iron, may influence this process by promoting the formation of the damaging agents, while other components, such as some flavonoids and folate, may function to enhance repair mechanisms[23]. For example, aqueous fractions of Fushimi sweet pepper have been reported to increase repair against ultraviolet-induced cyclobutane pyrimidine dimers in human fibroblasts[24] and kiwifruit consumption increased DNA repair capacity in human lymphocytes[25]. Other data point to the essentiality of folate in maintaining normal DNA synthesis and repair[26,27]. Dietary components that scavenge activated oxygen species, such as flavonoids, vitamins E and C and isothiocyanates have been shown to stimulate repair of oxidative DNA damage. Moreover, it has been shown that dietary supplementation with cooked carrots increased the repair of 8-oxodG (an indicator of oxidative DNA damage) in white blood cells, whereas a similar amount of α-carotene and β-carotene provided as capsules had no effect[28]. Some dietary components may also retard repair as has been suggested following alcohol exposure[29].
Cellular proliferation and death DNA damage can arrest cell cycle progression to allow for repair and the prevention of the replication of the defect or to activate apoptosis (programmed cell death) to eliminate cells with catastrophic mutations[30]. Cell homeostasis is regulated by a delicate balance of proliferation, growth arrest, differentiation and apoptosis. Alterations in DNA repair, cell cycle progression and apoptosis are all important molecular targets for dietary components in cancer prevention.
In general, the growth rate of pre-neoplastic or neoplastic cells outpaces that of normal cells because of malfunctioning or dysregulation of their cell-growth and cell-death machineries[31]. Cell cycle progression is a sequential process that directs dividing mammalian cells through G1, S, G2 and M phases. Transitions between G1-S or G2-M phases function as checkpoints to halt cell division if necessary. Because the balance of interactions among cyclins, cyclin-dependent kinases (CDK) and CDK inhibitors (CDI) governs the progression of the cell cycle[32], perturbation of any of the cell-cycle-specific proteins by dietary components can potentially affect and block the continuous proliferation of neoplastic cells and may serve as effect biomarkers. Dietary components that modulate cell proliferation include phenolic compounds, such as genistein and epigallocat-echin-3-gallate, which elicit cell-cycle arrest through the induction of CDI (p21 and p27) and the inhibition of CDK4, CDK2, cyclin D1 and cyclin E[33]. Isothiocyanates can also induce p21 expression and inhibit cell proliferation at the G2-M checkpoint[34]. Allyl sulfur compounds from allium foods have been reported to block the cell cycle in the G2/M phase presumably by inhibiting p34 (cdc2) kinase activity through changes in cyclin complex formation and hyperphosphorylation[35].
Apoptosis is one of the most potent defenses against cancer because this process eliminates potentially deleteri-ous, mutated cells. Many dietary cancer preventive compounds, including selenium, epigallocatechin-3-gallate, phenylethyl isothiocyanate, retinoic acid, sulforaphane, curcumin, apigenin, quercetin and resveratrol, induce apoptosis[36,37]. Distinct from the apoptotic events in the normal physiological process, which are mediated mainly by the interaction between death receptors and their relevant ligands[38], many bioactive dietary components appear to induce apoptosis through the mitochondria-mediated pathway. Dietary compounds generally induce oxidative stress, which downregulates anti-apoptotic molecules, such as Bcl-2 or Bcl-x, and upregulates pro-apoptotic molecules, such as Bax or Bak[39]. The imbalance between anti-apoptotic and pro-apoptotic proteins elicits the release of cytochrome c from the mitochondrial membrane, which forms a complex with caspase-9 with the subsequent activation of caspases 3, 6 and 7[40]. The activated caspases degrade important intracellular proteins, leading to morphological changes and the phenotype of the apoptotic cells[41]. To enhance this mitochondria-mediated apoptosis, dietary components also activate pro-apoptotic c-Jun N-terminal kinase (JNK) and inhibit anti-apoptotic NF-κB signaling[39]. Thus, potential “effect” biomarkers for the cytotoxic effect of dietary components on cells may be assessed by measuring their influence on mitochondrial caspases and other apoptosis-related proteins. Collectively, common cell cycle and apoptotic protein targets (biomarkers) relevant to the pathogenesis of cancer may be useful for not only diagnosis, but also in the development of preventive strategies, including dietary change.
Inflammation and immunonutrition The immune system represents a primary defense against invading patho-gens, non-self components and cancer cells. Inflammation is a basic process by which the body reacts to infection, irritations or other injuries and is recognized as a type of non-specific immune response. The inflammatory processes, including the release of pro-inflammatory cytokines [eg tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-6, IL-12, and γ-interferon] and the formation of reactive oxygen and nitrogen species, are critical factors driving this process and can be influenced by several dietary components. Although pro-inflammatory actions are usually followed almost immediately by anti-inflammatory responses (eg IL-4, IL-10 and TGF-β), excessive production of pro-inflammatory cytokines may lead to chronic inflammation. Evidence exists that selected dietary components including conjugated linoleic acid, long-chain omega-3 fatty acids such as those in fish oil, butyrate, epigallocatechin-3- gallate, curcumin, resveratrol, genistein, luteolin, quercetin, and vitamins A and D may influence unique molecular targets associated with the inflammatory process[42–47].
The immune system protects against infection by producing specific antibodies in response to antigens. Vaccine-specific serum antibody production, delayed-type hypersensitivity response, vaccine-specific or total secretory IgA in saliva, and the response to attenuated pathogens are classic markers that can be influenced by dietary habits. Markers including natural killer cell cytotoxicity, oxidative burst of phagocytes, lymphocyte proliferation and the cytokine pattern produced have also surfaced as potential predictors of immunocompetence. As no single marker permits firm conclusions about the ability of diet to modulate the entire immune system, combining markers appears to be a suitable strategy. It is also clear that excesses of some nutrients can enhance the immune system, while other food components can have detrimental effects[48].
Angiogenesis Angiogenesis, the development of new blood vessels from endothelial cells, is a crucial process in tumor pathogenesis because it sustains malignant cells with nutrients and oxygen[49]. During angiogenesis, endothelial cells are stimulated by various growth factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), and are attracted to the site where the new blood supply is needed by inflammatory cytokines and chemoattractants[50,51]. Chemotactic migration along this gradient is, however, possible only through the degradation of extracellular matrix components[52]. This is accomplished via matrix metalloproteinases (MMP). Preventing the expansion of new blood vessel networks results in reduced tumor size and metastasis and is another mechanism whereby dietary components inhibit tumor growth. Dietary components that inhibit angiogenesis include polyunsaturated fatty acids[53], flaxseed[54] and polyphenols such as epigallocate-chin-3-gallate, resveratrol, curcumin and genistein[55–57]. Recently, sphingosine 1-phosphate (S1P), a bioactive sphingolipid metabolite abundantly stored in platelets and released upon activation, has emerged as a potent, specific and selective endothelial cell chemoattractant. Red grape skin polyphenolic extract prevents and inhibits angiogenesis in the Matrigel model, decreases the basal motility of endothelial and cancer cells, and reverses the chemotactic effect of S1P and VEGF on bovine aortic endothelial cells, as well as the chemotactic effect of conditioned medium on human HT-1080 fibrosarcoma, human U-87 glioblastoma and human DAOY medulloblastoma cells[58]. Inhibition of S1P-induced and VEGF-induced endothelial cell chemotaxis by red grape skin polyphenols correlates with a decrease in early platelet-activating factor synthesis[58].
Summary of molecular targets Collectively, overwhelming evidence demonstrates that a variety of nutrients can influence a number of key intracellular targets that are associated with the cancer process. A fundamental action of several bioactive food components is that they serve as regulators of gene expression and/or modulate gene products. Transcriptomic or microarray analysis can provide clues about the mechanisms that underlie the beneficial or adverse effects of dietary components. Such analysis can identify important genes and related events that are altered in the pre-disease state and may, therefore, serve as molecular biomarkers and/or assist in identifying and characterizing the basic molecular pathways influenced by food components. Recently, gene expression changes in human leukocytes after consumption of high-protein or high-carbohydrate breakfasts demonstrated the potential of gene-expression profiling in blood to study the effects of dietary exposure in human intervention studies[59].
Typically, increasing intensity and duration of the exposure to dietary components increases the number of gene expressions that are modified[60,61]. Thus, dose and duration of exposure become fundamental considerations in interpreting findings from microrarray studies. In addition, although most studies are simple snapshots of genomic expression changes that can help identify important possible targets, they must be interpreted cautiously because of inherent biological variability[62]. Determining which one of the targets is most important in altering tumor growth will not be a simple task. Likewise, unraveling the multitude of interactions among nutrients with these key events makes the challenge even more daunting. Finally, inter-individual differences probably reflecting genetic polymorphisms can mask the response to a nutrient and thereby complicate this undertaking to an even greater extent. Nevertheless, deciphering the role of diet is fundamental to optimizing health and preventing disease. Access to this information should help resolve the inconsistencies within the literature and provide clues to strategies that may be developed to assist individuals in improving their health.
Susceptibility biomarkers
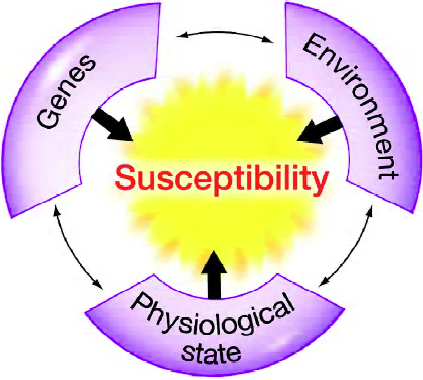
Complex gene–environment (including diet) and nutrient–nutrient interactions are also risk determinants for most disease states. Furthermore, an individual’s physiological state, such as the stage of the lifespan, will also influence their disease risk. Thus, an individual’s genes, environmental exposures and physiological state must all be considered when determining disease risk (Figure 4). The use of “susceptibility” biomarkers should assist in developing predictions about whether an individual, or animal, is more likely to be more sensitive than most other members of a population. Many of the differences in sensitivity may reflect variation in genetic backgrounds, namely genetic polymorphisms.
Gene–nutrient interactions The Human Genome Project suggests that approximately 30 000 genes exist. Most genes have base sequence differences as a result of single nucleotide polymorphisms (SNP), insertions and repeats. Some of the polymorphisms that occur in a gene coding sequence are of particular interest because they may cause an amino acid substitution and hence alter the biological function of a protein. For example, a SNP in the human gene encoding methylenetetrahydrolate reductase (MTHFR) results in the substitution of T for C at codon 677 and, thus, a substitution for alanine with valine. This substitution leads to reduced conversion of 5,10-methylenetretrahydrofolate to 5-methyl-enetetrahydrofolate, the form of folate that circulates in the plasma. Individuals with this polymorphism appear to have increased dietary folate and riboflavin requirements[63]. Several epidemiological studies have looked at SNP frequency and folate status in relation to colorectal cancer risk and most, but not all, suggest that the TT genotype is associated with decreased risk, particularly in combination with high dietary folate[reviewed in64].
Genomic data for human and mouse (including SNP, expressed sequence tags, gene expression patterns and cluster assemblies) and cytogenetic information are increasingly available through a number of databases; these databases provide opportunities to evaluate genomics as a factor in explaining variation in response to food components in terms of human growth, development, performance and health. Scanning the entire human genome for genetic variations should provide clues about human evolution. More than 700 genetic variants have already been identified and may have been targets of natural positive selection over the past 10 000 years and may reflect adaptations to various conditions, including the availability of a variety of foods and/or stress conditions. Examples of these websites include: www.ncgr.org; www.jgi.doe.gov; www.gmod.org; www.genome.gov; www.ebi.ac.uk; and www.nugo.org.
Increasingly, genetic polymorphisms are thought to have a role in determining the response to foods and their com-ponents. Unfortunately, while this area is receiving increased attention, it remains unclear whether these polymorphisms are directly linked to functional outcomes and disease risk. Nevertheless, it is certainly plausible that polymorphic differences have contributed to the inconsistencies among studies of the health effects of dietary components. In a random sample of participants in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC Study), for example, the low prevalence of polymorphisms in genes coding for activation (phase I) enzymes CYP1A1 (0.07) and CYP2E1 (0.02) and the high prevalence in genes coding for detoxification (phase II) enzymes GSTM1 (0.40) and NQO1 (0.20)[65] may have influenced the outcomes and conclusions of the study. Furthermore, in a nested case-control study within the ATBC Study, glutathione peroxidase 1 (hGPX1), a selenium-dependent enzyme involved in the detoxification of hydrogen peroxide, was found to have a polymorphism exhibiting a proline to leucine replacement at codon 198. This polymorphism conferred a relative risk for lung cancer of 1.8 for heterozygotes and 2.3 for homozygous variants compared with homozygote wild types[66].
In addition to glutathione peroxidase, genetic polymorphisms in other anti-oxidant enzymes can influence cancer risk and the benefits of dietary factors for cancer prevention. Manganese superoxide dismutase (MnSOD) is the main antioxidant enzyme in mitochondria. An SNP at codon 16 in the mitochondrial targeting sequence encodes for an alanine rather than a valine in the protein. This amino acid change affects the secondary structure of the protein and the alanine-containing protein is more efficiently transported into the mitochondria[67]. Risk of both breast and prostate cancer appears to be increased by this polymorphism[68,69]. More-over, in the Physicians’ Health Study, high pre-diagnostic concentrations of several plasma antioxidants (ie carotenoids, vitamin E, retinol and selenium) were associated with a significant reduction in aggressive prostate cancer risk only in individuals containing the alanine-containing protein[70].
Catalase is another important anti-oxidant enzyme. A polymorphism in the promoter region leads to the substitution of T for A at codon 262. This polymorphism is associated with a dose-response reduction in catalase activity (115.4, 82.1 and 73.5 units/mg hemoglobin) in the CC, CT and TT genotypes, respectively[71]. The high activity catalase genotype (CC) was also associated with a 17% reduction in the risk of breast cancer compared with having at least one variant T allele[71]. Moreover, differences in catalase activity by genotype were most pronounced among those in the highest tertiles of consumption of fruits and vegetables[71]. These types of studies suggest that some, but possibly not all, individuals because of genetic polymorphisms in anti-oxidant enzymes may be at increased risk for cancer and may benefit most from increased fruit and vegetable con-sumption.
Thus far, nutrition research has focused mainly on a few SNP at a time and on the opportunity to modify the consequences of a SNP, either positively or negatively, by nutritional intervention. Developments in genotyping technology make it possible to ascertain hundreds of thousands of variants in a single sample of DNA. For example, microarrays containing around 500 000 SNP (500 K arrays) are already available. These chips are being used in association studies, such as the Cancer Genetic Markers of Susceptibility (CGEMS) Project, to identify candidate loci for cancer and have identified common variants on human chromosome 8q24 that are associated with prostate cancer risk[72].
Literally millions of SNP occur within the human genome making it unlikely that a single base change will be found that is sufficient to account for a number of chronic diseases. However, because genetic variants are often inherited together in segments of DNA called haplotypes, which are shared by a majority of the human population, they may be useful in deciphering the genetic differences that make some people more susceptible to disease than others, and likewise how diet will impact their susceptibility. The International HapMap Project (http://www.hapmap.org/) may be particularly useful in teasing out genetic differences that determine the response to specific foods and their components.
Epigenetic modification can modify gene expression and disease susceptibility. Evidence already exists that transcriptional silencing of genes by DNA methylation plays a crucial role in a number of disease states[73,74]. Genes involving cell cycle regulation, DNA repair, angiogenesis and apoptosis are all inactivated by the hypermethylation of their respective 5’CpG islands. Key regulatory genes – including E-cadherin, pi-class glutathione S-transferase, the tumor suppressors cyclin-dependent kinases (CDKN2) and phosphatase gene (PTEN), and insulin-like growth factor (IGF-II) targeted histone acetylation and deacetylation – are influenced by DNA hypermethylation. Although folate intake is recognized to influence DNA methylation patterns, other nutrients, such as selenium, can also have an impact[75]. Restoring proper methylation may represent a fundamental process by which selected nutrients are able to influence gene expression. Epigallocatechin-3-gallate, the major polyphenol from geen tea, can inhibit DNA methyltransferase activity and reactivate methylation-silenced genes in cancer cells[76]. Recently, Fang et al[77] have provided evidence that indicates that feeding genistein and related soy isoflavones can also reactivate methylation-silenced genes, partially through the direct inhibition of DNA methyltransferase. These findings need confirmation and the impact of other dietary constituents in re-establishing normal methylation patterns deserves additional attention.
Alterations in mRNA, protein and metabolite expression may also be considered to be susceptibility factors. For example, if inhibition of a specific molecular target for a bioactive dietary component is the desired biological response and an individual has increased expression of the molecular target, than they may need more of the dietary component to bring about the desired biological effect. Thus, transcriptomics, proteomics and metabolomics may provide information about individual susceptibility.
Nutrient–nutrient interactions Interactions among dietary components may affect “susceptibility” by modifying the dose of nutrients that are required to bring about a physiological effect. This is exemplified by the inability of low doses of 9-cis-retinoic acid and vitamin D3 to prevent mammary cancer when given alone, but when given in combination they were effective[78]. Furthermore, the lower dosages bypassed potential adverse effects of higher intakes[78]. The mechanism responsible for this interaction is not known, but may involve binding of RAR, RXR and VDR, which could regulate the genes involved with cell proliferation, differentiation and/or apoptosis. Similarly, low doses of S-allyl-cysteine and lycopene in combination were able to suppress the development of MNNG-induced gastric cancer via modulation of apoptosis-associated proteins (decreased Bcl-2/Bax ratio and upregulation of Bim and caspases 8 and 3) at much lower intakes than when given in isolation[79]. Finally, the combination of vitamin D3 and genistein caused growth inhibition of DU145 prostate cancer cells at lower, and biologically achievable, concentrations compared with either compound alone[75]. Genistein appears to potentiate the action of vitamin D3 by directly inhibiting CYP24 enzyme activity and, therefore, increasing the half-life of vitamin D3, which results in homologous upregulation of cellular VDR levels[80]. This dual action of genistein leads to enhanced vitamin D-mediated responses and target gene activation renders the cells more sensitive to the growth inhibitory and pro-apoptotic signals of vitamin D3[80].
Dietary components that alter multiple molecular targets within a specific biological process or pathway may exert additive or synergistic effects. For example, dietary components that inhibit different phases of the cell cycle cooperate to inhibit tumor growth. Quercetin and genistein synergistically inhibit proliferation of ovarian carcinoma cells by modifying different stages in the cell cycle and different signal transduction pathways[81]. Quercetin arrests the cell cycle at the G1 and S phase boundary, whereas genistein affects the G2 and/or early M phase. Similarly, quercetin interacts synergistically with resveratrol to cause transient cell cycle arrest in human leukemia cells[82] and epigallocatechin-3-gallate and curcumin synergistically inhibit the growth of normal, premalignant and malignant human oral epithelial cells[83]. Whereas EGCG blocked cells in G1, curcumin blocks cells in S/G2M. Thus, synergistic interactions between/among dietary phytochemicals are likely to contribute to inhibition of cell proliferation.
A critical unanswered question is what additive or antagonistic responses occur among dietary components that have the same molecular target. For example, organosulfur compounds in garlic and sulforaphane in broccoli can both induce expression of detoxifying enzymes via the binding of the transcription factor Nrf2 to the anti-oxidant response element (ARE). It remains unclear if broccoli would illicit protective effects if Nrf2 was maximized by consumption of organosulfur compounds. Additional information is needed to determine the biological significance of nutrient–nutrient interactions and their influence on susceptibility biomarkers.
Physiological state Cancer susceptibility can also be modified by the timing and duration of exposure to dietary components. Despite its well-recognized limitations, birth weight has been used widely as a summary measure of the normality of intrauterine growth[84]. There is evidence that higher birth weight may increase the overall risk of cancer in adulthood in both men and women[85]. Thus, individuals who are born overweight may have increased susceptibility regardless of genetics or future environmental exposures.
The timing of dietary exposure may also influence susceptibility. In a rat model of carcinogen-induced mammary cancer, limiting exposure to dietary genistein, the primary isoflavone of soy, to the prenatal or adult periods does not predispose or protect against mammary cancer[86]. In contrast, exposure to dietary genistein during the prepubertal and prepubertal plus adult periods protected against mammary cancer[86]. Similarly, a case-control study in Shanghai has shown an inverse relationship between adolescent 13–15-year olds, soy food intake and breast cancer incidence later in life[87]. These data suggest that the consumption of soy during the prepubertal period, compared to other stages of the life cycle, may be more efficacious against mammary cancer development.
Another important consideration is whether the effects of dietary components persist for extended periods. Results from the General Population Trial in Linxian, China, demonstrated that individuals who received a supplement containing beta-carotene, vitamin E and selenium, had a 13% reduction in cancer mortality. Post-intervention follow up indicated that the beneficial effects of the supplement were still evident up to 10 years after termination of the supplementation[88]. Moreover, these benefits were consistently greater in participants who were younger (<55 years) at the beginning of the intervention and cancer risk appeared to increase in those who started supplement use after the age of 55 years compared with those who started supplement use when they were <55 years[88]. Evidence for a carry-over effect of nutrients also comes from observations made in a cohort study in Washington County, Maryland, USA. In this study an inverse relationship between circulating 25(OH)D, a biomarker of vitamin D exposure, and colon cancer was observed in the first 8 years after the blood sample collection, but no association was observed in cases diagnosed 10–17 years after the sample collection[89]. Results such as these suggest that an individual may still have decreased cancer susceptibility after discontinuing exposure to beneficial dietary components, but the positive effects are unlikely to last indefinitely. Regardless, a better understanding of the temporal relationship between exposure to a dietary component, whether as foods or supplements, warrants additional attention.
For most cancers, increased age is a significant susceptibility factor[90]. The cancer-prone phenotype of older humans may reflect the combined effects of accumulation of DNA damage, increased epigenetic gene silencing, telomere dysfunction and altered stromal milieu[90]. Because most cancers occur during the later stages of life, a better understanding of the age dependency of diet–genetic interactions is needed.
The response to dietary components may also depend on the presence of preneoplastic lesions or tumors at the time of intervention. Animal studies using chemical and genetically predisposed models provide strong evidence for a causal relationship between folate depletion and increased cancer incidence, as well as a dose-dependent protective effect of folate supplementation[reviews in 91]. However, animal studies have also shown that the dose and timing of folate intervention are critical in providing safe and effective cancer prevention[91]. Exceptionally high supplemental folate levels and folate intervention after microscopic neoplastic foci are established in the colorectal mucosa can promote rather than suppress colorectal carcinogensis[91]. Such evidence suggests that the optimal dose of folate for cancer prevention may vary among individuals and may depend on whether or not microscopic lesions are present.
Summary
Dietary modifications and interventions have the potential to significantly lower cancer risk and its associated complications. Validated biomarkers would be invaluable tools to facilitate research in this area and to identify those who would respond to dietary change. Biomarkers of exposure would allow evaluation as to whether sufficient intakes are achieved to bring about a biological response in a particular cellular process. Biomarkers of biological effect will provide insights into sites of action and, thus, mechanisms of action of dietary components. In addition, biomarkers are needed to identify individuals susceptible to specific dietary exposures. Despite promising recent research, there remains an enormous need for defining the applicability of non-invasive biomarkers in humans. The application of molecular genetics is likely to assist in achieving greater precision in identifying responsive individuals. It is very likely that a suite of biomarkers, rather than a single measure, will be needed to adequately evaluate the impact of altering dietary intakes on overall health and disease prevention.
References
- World Cancer Research Fund, American Institute for Cancer Research. Food, Nutrition and the Prevention of Cancer: a Global Perspective. Washington: American Institute for Cancer Research; 1997.
- Milner JA. Molecular targets for bioactive food components. J Nutr 2004;134:2492s-8s.
- Liu RH. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am J Clin Nutr 2003;78:517S-20S.
- Davis CD. Nutritional interactions: credentialing of molecular targets for cancer prevention. Exp Biol Med 2007;232:176-83.
- Coates PM, Milner JA. Bioactive components in foods and supplements for health promotion. In: Bowman BA, Russel RA, editors. Present knowledge in nutrition; 9th ed. Washington: ILSI; 2006. p 959–67.
- Timbrell JA. Biomarkers in toxicology. Toxicology 1998;129:1-12.
- Suk WA, Collman GW. Genes and the environment: their impact on children’s health. Environ Health Perspect 1998;106:817-20.
- Vainio H. Use of biomarkers — new frontiers in occupational toxicology and epidemiology. Toxicol Lett 1998;102–103:581-9.
- Davis CD, Milner J. Frontiers in nutrigenomics, proteomics, metabolomics and cancer prevention. Mutat Res 2004;551:51-64.
- Davis CD. Nutrigenomics and the prevention of colon cancer. Pharmacogenomics 2007;8:121-4.
- Trujillo E, Davis CD, Milner J. Nutrigenomics, proteomics, metabolomics and the practice of dietetics. J Am Diet Assoc 2006;106:403-13.
- Dodd KW, Guenther PM, Freedman LS, Subar AF, Kipnis V, Midthune D, et al. Statistical methods for estimating usual intake of nutrients and foods: a review of the theory. J Am Diet Assoc 2006;106:1640-50.
- Olafsdottir AS, Thorsdottir I, Gunnarsdottir I, Thorgeirsdottir H, Steingrimsdottir L. Comparison of women’s diet assessed by FFQs and 24-hour recalls with and without underreporters: associations with biomarkers. Ann Nutr Metab 2006;50:450-60.
- Guthrie HA, Crocetti AF. Variability of nutrient intake over a 3-day period. J Am Diet Assoc 1985;85:325-7.
- Michels KB, Bingham SA, Luben R, Welch AA, Day NE. The effect of correlated measurement error in multivariate modules of diet. Am J Epidemiol 2004;160:59-67.
- Subar AF, Dodd KW, Guenther PM, Kipnis V, Midthune D, McDowell M, et al. The food propensity questionnaire: concept, development, and validation for use as a covariate in a model to estimate usual food intake. J Am Diet Assoc 2006;106:1556-63.
- McKeown NM, Day NE, Welch AA, Runswick SA, Luben RN, Mulligan AA, et al. Use of biological markers to validate self-reported dietary intake in a random sample of the European Prospective Investigation into Cancer United Kingdom Norfolk cohort. Am J Clin Nutr 2001;74:188-96.
- Finley JW. Selenium accumulation in plant foods. Nutr Rev 2005;63:196-202.
- Kushad MM, Brown AF, Kurilich AC, Juvik JA, Klein BP, Wallig MA, et al. Variation of glucosinolates in vegetable crops of Brassica oleracea. J Agr Food Chem 1999;47:1541-8.
- de Vries JH, Hollman PC, Meyboom S, Buysman MN, Zock PL, van Staveren WA, et al. Plasma concentrations and urinary excretion of the antioxidant flavonols quercetin and kaempferol as biomarkers for dietary intake. Am J Clin Nutr 1998;68:60-5.
- Stahl W, Sies H. Uptake of lycopene and its geometrical isomers is greater from heat-processed than from unprocessed tomato juice in humans. J Nutr 1992;122:2161-6.
- Song K, Milner JA. Heating garlic inhibits its ability to suppress 7,12-dimethylbenz(a)anthracene-induced DNA adduct formation in rat mammary tissue. J Nutr 1999;129:657-61.
- Fenech M. The Genome Health Clinic and Genome Health Nutrigenomics concepts: diagnosis and nutritional treatment of genome and epigenome damage on an individual basis. Mutagenesis 2005;20:255-69.
- Nakamura Y, Tomokane I, Mori T, Tanaka A, Koutani J, Matsui T, et al. DNA repair effect of traditional sweet pepper Fushimi-togarashi: seen in suppression of UV-induced cyclobutane pyrimidine dimer in human fibroblast. Biosci Biotechnol Biochem 2000;64:2575-80.
- Freese R. Markers of oxidative DNA damage in human interventions with fruits and berries. Nutr Cancer 2006;54:143-7.
- Beetstra S, Thomas P, Salisbury C, Turner J, Fenech M. Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. Mutat Res 2005;578:317-26.
- Friso S, Choi SW. Gene–nutrient interactions in one-carbon metabolism. Curr Drug Metab 2005;6:37-46.
- Astley SB, Elliott RM, Archer DB, Southon S. Evidence that dietary supplementation with carotenoids and carotenoid-rich food modulates the DNA damage: repair balance in human lymphocytes. Br J Nutr 2004;91:63-72.
- Hong YC, Lee KH, Kim WC, Choi SK, Woo ZH, Schin SK, et al. Polymorphisms of XRCC1 gene, alcohol consumption and colorectal cancer. Int J Cancer 2005;116:428-32.
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004;73:39-85.
- Jacks T, Weinberg RA. Taking the study of cancer cell survival to a new dimension. Cell 2002;111:923-5.
- Weinstein IB. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis 2000;21:857-64.
- Agarwal R. Cell signaling and regulators of cell cycle as molecular targets for prostate cancer prevention by dietary agents. Biochem Pharmacol 2000;60:1051-9.
- Visanji JM, Duthie SJ, Pirie L, Thompson DG, Padfield PJ. Dietary isothiocyanates inhibit Caco-2 cell proliferation and induce G2/M phase cell cycle arrest, DNA damage, and G2/M checkpoint activation. J Nutr 2004;134:3121-6.
- Knowles LM, Milner JA. Diallyl disulfide inhibits p34(cdc2) kinase activity through changes in complex formation and phosphorylation. Carcinogenesis 2000;21:1129-34.
- Martin KR. Targeting apoptosis with dietary bioactive agents. Exp Biol Med 2006;231:117-29.
- Hu R, Kong AN. Activation of MAP kinases, apoptosis and nutrigenomics of gene expression elicited by dietary cancer-prevention compounds. Nutrition 2004;20:83-8.
- Krammer PH. CD95’s deadly mission in the immune system. Nature 2000;407:789-95.
- Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol Sci 2005;26:318-26.
- Watson RW, Fitzpatrick JM. Targeting apoptosis in prostate cancer: focus on caspases and inhibitors of apoptosis proteins. BJU Int 2005;96:30-4.
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science 1988;281:1312-6.
- Caughey GE, Mantzioris E, Gibson RA, Cleland LG, James MJ. The effect on human tumor necrosis factor alpha and interleukin 1 beta production of diets enriched in n-3 fatty acids from vegetable oil or fish oil. Am J Clin Nutr 1996;63:116-22.
- Shany S, Levy Y, Lahav-Cohen M. The effects of 1alpha,24(S)-dihydroxyvitamin D(2) analog on cancer cell proliferation and cytokine expression. Steroids 2001;66:319-25.
- Chan MM. Inhibition of tumor necrosis factor by curcumin, a phytochemical. Biochem Pharmacol 1995;49:1551-6.
- Xagorari A, Papapetropoulos A, Mauromatis A, Economou M, Fotsis T, Roussos C. Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J Pharmacol Exp Ther 2001;296:181-7.
- Davis JN, Kucuk O, Djuric Z, Sarkar FH. Soy isoflavone supplementation in healthy men prevents NF-kappaB activation by TNF-alpha in blood lymphocytes. Free Radic Biol Med 2001;30:1293-302.
- Manna SK, Aggarwal BB. All-trans-retinoic acid upregulates TNF receptors and potentiates TNF-induced activation of nuclear factor-kappaB, activated protein-1 and apoptosis in human lung cancer cells. Oncogene 2000;19:2110-9.
- Calder PC, Kew S. The immune system: a target for functional foods? Br J Nutr 2002;88 Suppl 2:S165-77.
- Fayette J, Soria JC, Armand JP. Use of angiogenesis inhibitors in tumour treatment. Eur J Cancer 2005;41:1109-16.
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005;16:159-78.
- Albini A, Tosetti F, Benelli R, Noonan DM. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res 2005;65:10637-41.
- Pfeffer U, Ferrari N, Morini M, Benelli R, Noonan DM, Albini A. Antiangiogenic activity of chemopreventive drugs. Int J Biol Markers 2003;18:70-4.
- Rose DP, Connolly JM. Regulation of tumor angiogenesis by dietary fatty acids and eicosanoids. Nutr Cancer 2000;37:119-27.
- Bergman Jungestrom M, Thompson LU, Dabrosin C. Flaxseed and its lignans inhibit estradiol-induced growth, angiogenesis, and secretion of vascular endothelial growth factor in human breast xenografts in vivo. Clin Cancer Res 2007;13:1061-7.
- Cao Y, Cao R, Brakenhielm E. Angioangiogenic mechanisms of diet-derived polyphenols. J Nutr Biochem 2002;13:380-90.
- Dulak J. Nutraceuticals as anti-angiogenic agents: hope and reality. J Physiol Pharmacol 2005;56:51-69.
- Oak MH, El Bedoui J, Schini-Kerth VB. Antioangiogenic properties of natural polyphenols from red wine and green tea. J Nutr Biochem 2005;16:1-8.
- Barthomeuf C, Lamy S, Blanchette M, Boivin D, Gingras D, Beliveau R. Inhibition of sphingosine-1-phosphate- and vascular endothelial growth factor-induced endothelial cell chemotaxis by red grape skin polyphenols correlates with a decrease in early platelet-activating factor synthesis. Free Radic Biol Med 2006;40:581-90.
- van Erk MJ, Blom WA, van Ommen B, Hendriks HF. High-protein and high-carbohydrate breakfasts differentially change the transcriptome of human blood cells. Am J Clin Nutr 2006;84:1233-41.
- El-Bayoumy K, Sinha R. Molecular chemoprevention by selenium: A genomic approach. Mutat Res 2005;591:224-36.
- Prima V, Tennant M, Gorbatyuk OS, Muzyczka N, Scarpace PJ, Zolotkhin S. Differential modulation of energy balance by leptin, ciliary neurotrophic factor, and leukemia inhibitory factor gene delivery: microarray deoxyribonucleic acid-chip analysis of gene expression. Endocrinology 2004;145:2035-45.
- Zakharkin SO, Kim K, Mehta T, Chen L, Barnes S, Scheirer KE, et al. Sources of variation in Affymetrix microarray experiments. BMC Bioinformatics 2005;6:214.
- van den Donk M, Buijsse B, van den Berg SW, Ocké MC, Harryvan JL, Nagengast FM, et al. Dietary intake of folate and riboflavin, MTHFR C677T genotype, and colorectal adenoma risk: a Dutch case-control study. Cancer Epidemiol Biomarkers Prev 2005;14:1562-6.
- Ulrich CM, Curtin K, Potter JD, Bigler J, Caan B, Slattery ML. Polymorphisms in the reduced folate carrier, thymidylate synthase, or methionine synthase and risk of colon cancer. Cancer Epidemiol Biomarkers Prev 2005;14:2509-16.
- Woodson K, Ratnasinghe D, Bhat NK, Stewart C, Tangrea JA, Hartman TJ, et al. Prevalence of disease-related DNA polymorphisms among participants in a large cancer prevention trial. Eur J Cancer Prev 1999;8:441-7.
- Ratnasinghe D, Tangrea JA, Andersen MR, Barrett MJ, Virtamo J, Taylor PR, et al. Glutathione peroxidase codon 198 polymorphism variant increases lung cancer risk. Cancer Res 2000;60:6381-3.
- Sutton A, Khoury H, Prip-Buus C, Cepanec C, Pessayre D, Degoul F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver miotchondria. Pharmacogenetics 2003;13:145-57.
- Ambrosone CB, Freudenheim JL, Thompson PA, Bowman E, Vena JE, Marshall JR, et al. Manganese superoxide dismutase (MnSOD) genetic polymorphisms, dietary antioxidants, and risk of breast cancer. Cancer Res 1999;59:602-6.
- Woodson K, Tangrea JA, Lehman TA, Modali R, Taylor KM, Snyder K, et al. Manganese superoxide dismutase (MnSOD) polymorphism, alpha-tocopherol supplementation and prostate cancer risk in the alpha-tocopherol, beta-carotene cancer prevention study (Finland). Cancer Causes Control 2003;14:513-8.
- Li H, Kantoff PW, Giovannucci E, Leitzmann MF, Gaziano JM, Stampfer MJ, et al. Manganese superoxide dismuatase polymorphism, prediagnostic antioxidant status, and risk of clinical significant prostate cancer. Cancer Res 2005;65:2498-504.
- Ahn J, Nowell S, McCann SE, Yu J, Carter L, Lang NP, et al. Associations between catalase phenotype and genotype: modification by epidemiological factors. Cancer Epidemiol Biomarkers Prev 2006;15:1217-22.
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007;39:645-9.
- Ross SA. Diet and DNA methylation interactions in cancer prevention. Ann NY Acad Sci 2003;983:197-207.
- Davis CD, Uthus EO. DNA methylation, cancer susceptibility and nutrient interactions. Exp Biol Med (Maywood) 2004;229:998-5.
- Davis CD, Uthus EO. Dietary folate and selenium affect dimethylhydrazine-induced aberrant crypt formation, global DNA methylation and one-carbon metabolism in rats. J Nutr 2003;133:2907-14.
- Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003;63:7563-70.
- Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res 2005;11:7033-41.
- Cope MB, Steele VE, Eto I, Juliana MM, Grubbs CJ. Prevention of methylnitrosourea-induced mammary cancers by 9-cis-retinoic acid and/or vitamin D3. Oncol Rep 2002;9:533-7.
- Velmurugan B, Mani A, Nagini S. Combination of S-allylcysteine and lycopene induces apoptosis by modulating Bcl-2, Bax, Bim and caspases during experimental gastric carcinogenesis. Eur J Cancer Prev 2005;14:387-93.
- Swami S, Krishnan AV, Peehl DM, Feldman D. Genistein potentiates the growth inhibitory effects of 1,25-dihydroxyvitamin D3 in DU145 human prostate cancer cells: role of the direct inhibition of CYP24 enzyme activity. Mol Cell Endocrinol 2005;241:49-61.
- Shen F, Weber G. Synergistic action of quercetin and genistein in human ovarian carcinoma cells. Oncol Res 1997;9:597-602.
- Mertens-Talcott SU, Percival SS. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett 2005;21:141-51.
- Khafif A, Schantz SP, Chou TC, Edelstein D, Sacks PG. Quantitation of chemopreventive synergism between (-)-epigallocatechin-3-gallate and curcumin in normal, premalignant and malignant human oral epithelial cells. Carcinogenesis 1998;9:419-24.
- Mathers JC, Hesketh JE. The biological revolution: understanding the impact of SNPs on diet–cancer interrelationships. J Nutr 2007;137:253s-8s.
- McCormack VA, dos Santos Silva I, Koupil I, Leon DA, Lithell HO. Birth characteristics and adult cancer incidence: Swedish cohort of over 11,000 men and women. Int J Cancer 2005;115:611-7.
- Lamartiniere CA, Cotroneo MS, Fritz WA, Wang J, Mentro-Mercel R, Elgavish A. Genistein chemoprevention: timing and mechanisms of action in murine mammary and prostate. J Nutr 2002;132:552s-8s.
- Boyapati SM, Xhu XO, Ruan ZX, Dai Q, Cai Q, Gao YT, et al. Soyfood intake and breast cancer survival: a followup of the Shanghai Breast Cancer Study. Breast Cancer Res Treat 2005;92:11-7.
- Taylor PR, Qiao Y, Dawsey SM, Johnson LL, Dong Z, Yu B, et al. Total and cancer mortality following supplementation with multi-vitamins and minerals: post-intervention follow-up of a general population nutrition intervention trial in Linxian China. Gastroenterology 2005;218 Suppl 2:A296.
- Braun MM, Helzlsouer KJ, Hollis BW, Comstock GW. Colon cancer and serum vitamin D metabolites levels 10–17 years prior to diagnosis. Am J Epidemiol 1995;142:608-11.
- DePinho RA. The age of cancer. Nature 2000;408:248-54.
- Kim YI. Folate, colorectal carcinogenesis, and DNA methylation: lessons from animal studies. Environ Mol Mutagen 2004;44:10-25.