
Cross-talk between oxidative stress and modifications of cholinergic and glutaminergic receptors in the pathogenesis of Alzheimer's disease1
Introduction
Alzheimer’s disease (AD) is the most common neuro-degenerative disorder of the elderly. The disease occurs both in sporadic and hereditary forms, and the main clinical features of this disease are progressive memory loss, decline in language skills, and other cognitive impairments[1]. The major pathological hallmarks in AD are senile plaques (SP) and neurofibrillary tangles (NFT), which have for the most part been regarded as central mediators of neuronal cell death leading to cognitive decline and eventual demise[2,3]. The core of SP is abundantly deposited with β-amyloid peptides (Aβ), a 39–43 amino acid peptide, which is the result of proteolytic cleavage of the transmembrane amyloid precursor protein (APP). The NFT contain paired helical filaments that are mostly composed of a hyperphosphorylated form of the microtubule-associated tau protein.
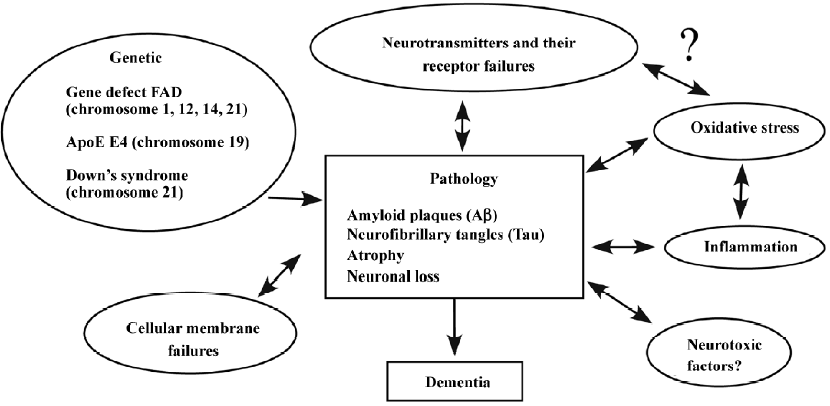
Although the initiating causes leading to AD are unknown, it is clear that its pathophysiology is complex and most likely involves multiple distinct and overlapping pathways of neuronal damage, including genetic deficit, oxidative stress, impaired neurotransmitter receptors, and other biological or biochemical abnormalities (Figure 1). Since the pathogenesis of AD remains elusive, there is no efficient cure of the disorder, and targeting the disruption of neurotransmission is the most viable therapeutic strategy at present[4].
Oxidative stress in AD
Oxidative stress, defined as a disturbance in the balance between the production of free radicals and antioxidative defense, may play an important role in several pathological conditions of the central nervous system (CNS)[5,6]. There is a great deal of evidence to suggest that the damage induced by free radicals may be an important pathogenesis in AD[7]. Free radicals, such as superoxide, hydroxyl ions, and nitric oxide, cause cell injury by damaging lipids, protein, and DNA when they are generated in excess or antioxidative defense is impaired[8]. The brain is more vulnerable to oxyradical-mediated injury because its cellular membranes are preferentially enriched in oxyradical-sensitive polyunsaturated fatty acids that are the substrates of lipid peroxidation, and damaged adult neurons cannot be replaced.
Multiple lines of evidence link oxidative stress and AD. In brain tissues from AD, carbonyls derived from protein oxidation were increased in the neuronal cytoplasm and nuclei of neurons and glial cells[9]. Moreover, NFT were strongly labeled by carbonyls. Transgenic animals with Aβ overexpression show the same type of oxidative damage found in AD, and this damage directly correlates with the presence of Aβ deposits[10]. To diagnose AD in clinical examinations, oxidative stress markers, such as 3-nitrotyrosine, 8-hydroxy-2'-deoxyguanosine, and isoprostanes, are found to be increased in cerebrospinal fluid in AD patients[11]. Interestingly, the elevation of lipid peroxidation in transgenic mice with APP mutations has been observed to precede the surge in the Aβ level and amyloid plaque formation[12,13].
Reduced glucose metabolism and mitochondrial abnormalities are associated with AD, as mitochondrial abnormalities are considered to be the source of oxidative stress in AD[14]. Mitochondrial abnormalities have been associated with deficiencies in enzyme activities, specifically the α-keto-glutarate dehydrogenase complex, pyruvate dehydrogenase complex, and cytochrome oxidase in AD neurons. Oxidative phosphorylation produces superoxide radicals as a byproduct associated with electron transport. Heightened superoxide radical formation in AD also correlates with heightened superoxide dismutase levels that may allow the release of H2O2 from the mitochondria to the cytoplasm[15].
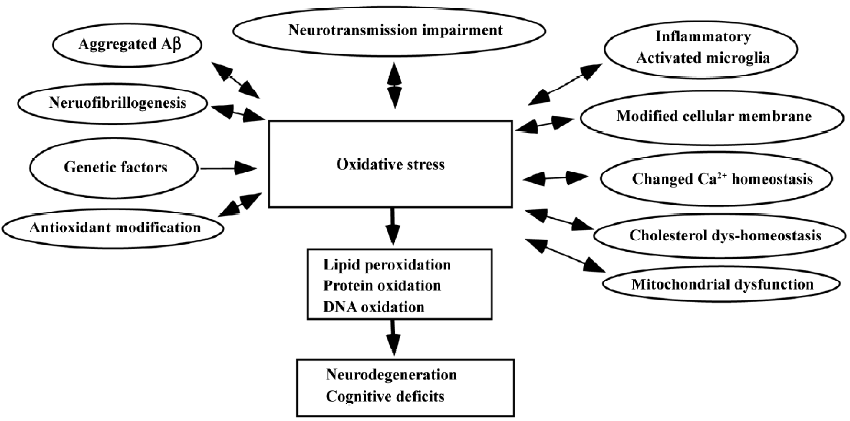
Recent studies of living patients and transgenic models of the disease have shed light on the central issue of whether oxidative stress is a result of the pathology of AD or whether it is an initiator of pathological damage. From our studies relating to oxidative stress in AD, we have shown decreased cellular membrane phospholipids and polyunsaturated fatty acids in the AD brain, in which these specific lipid modifications strongly support the involvement of free radicals in the pathogenesis of AD[16]. A closed correlation has been found between the high level of lipid peroxidation and the decreased number of nicotinic receptors in the AD brain[17]. We have suggested that oxidative stress is an early event in AD and is likely to play a more active role in the pathogenesis than previously hypothesized[18]. The mechanisms of oxidative stress in AD are shown in Figure 2.
Aggregated Aβ is toxic in a variety of neuronal cell preparations, and the overexpression of Aβ in transgenic mice leads to increased neuronal oxidative stress. Interestingly, recent evidence, however, has revealed that Aβ and NTF might have a compensatory response induced by reactive oxygen species (ROS) and have neurotrophic and potentially cytoprotective properties[19].
Changed cholinergic and glutaminergic receptors in AD
There is a great deal of evidence that suggests that in the AD brain, a number of neurotransmitter receptor systems are defective. Such abnormalities include cholinergic-, glutaminergic-, dopaminergic-, and γ-amino butyric acid-receptor (GABA) systems. Of all the neurotransmitter systems studied, cholinergic and glutaminergic receptors are more noticeably involved in the pathogenesis of AD.
Neuronal nicotinic acetylcholine receptors in AD
In cholinergic transmission, neuronal nicotinic acetylcholine receptors (nAChRs) have been extensively investigated during the progress of AD. The nAChRs belong to a gene super family of homologous receptors, including nicotinic, GABA, glycine, serotonin, and glutamate receptors[20]. The nAChRs in the brain are composed of 2 types of subunits, α and β. The gene family for neuronal nAChRs contains at least 9 α subunits (α2–α10) and 3 β subunits (β2–β4). Like the muscle type of nAChRs, the agonist recognition site of the neuronal nAChRs is on the interface between the α and β subunits. The expression of combinations of different α subunits with β subunits may produce functionally distinct neuronal nAChRs in different brain regions. The α4β2 and α7 subtypes are the most common patterns of nAChRs in the brain[20].
Interestingly, nAChRs are involved in many brain functions, such as cognition, memory, and neuroprotection, and a pathogenic role of the receptors has been established in AD[21]. Studies of AD patients with positron emission tomography have shown significant deficits in nAChRs early in the course of the disease, which is related to cognitive function. Significant reductions in the number of nAChRs were also observed in various autopsy brain cortical regions of the Swedish 670/671 APP mutation family[22]. In our previous investigations, a decreased level of nAChR subunit proteins, such as α4, α7, and α3, were detected in AD brains[23,24]. A large loss of the receptors in AD brains and in a cellular model provoked interest in developing therapeutic agonists for specific nAChR subtypes. In a recent study, we found that suppressed gene expression of nAChRs resulted from treatment by nanomolar Aβ25–35 and Aβ40 in PC12 cells[25].
The α7 nAChR is of importance in an AD study due to its neuroprotective function[26]. The α7 nAChR is highly expressed on neurons of the hippocampus and cholinergic projection neurons from the basal forebrain. A number of recent studies have convincingly demonstrated an interaction between the α7 nAChR and Aβ in vitro and on neurons. For instance, it was shown that Aβ1–42 co-immunoprecipitates with the α7 nAChR in the samples from a postmortem AD hippocampus, and Aβ can activate α7 nAChR[27]. A recent study has shown a selective increased α7 nAChR in APPSWE transgenic mice, which is not consistent with earlier changes in AD, and suggested important protective compensatory mechanisms in response to deficits in synaptic plasticity induced by elevated soluble Aβ[28]. We observed an increase in the α7 subunit in astrocytes and a decrease in the same receptor subtype in neurons in the hippocampus and the temporal cortex of AD patients who died carrying the APPSWE mutation[24]. The findings suggest different regulatory mechanisms and roles of the α7 nAChR in astrocytes and neurons.
Muscarinic acetylcholine receptors in AD
The muscarinic acetylcholine receptors (mAChRs) mediate most of the actions of the neurotransmitter acetylcholine in the CNS and peripheral nervous system, as well as in the end organs of parasympathetic nerves[29]. In mammals, 5 distinct mAChR subtypes (M1–M5) have been identified, with each receptor subtype being the product of a different gene. The mAChR belongs to the super family of 7 transmembrane receptors, which activate signal transduction pathways through their interaction with GTP binding proteins (G-proteins). mAChRs are stimulated by agonists, such as acetylcholine, which activates G-protein and evokes typically slow and modulatory second message responses. In cholinergic transmission, mAChRs have been implicated in higher brain functions, such as learning and memory, and the loss of muscarinic cholinergic synapses may contribute to the symptoms of AD[30].
Among the 5 types of mAChRs, the M1 mAChR is predominant in the cerebral cortex and hippocampus and has attracted great interest for its major function in cognitive processing relevant to AD, in particular short-term memory. An early study showed that a significantly reduced number of [3H] quinuclidinyl benzilate binding sites was obtained in AD dementia groups. A significant negative correlation was observed in AD brains between the histopathological dementia score and the reduction in the activity of the acetylcholine synthesizing enzyme choline acetyltransferase[31]. A disturbance of the muscarinic receptor–G-protein coupling in AD has been suggested, which might be impaired by the formation of Aβ. This can lead to decreased signal transduction, a decreased secreted form of APP, and increased generation of Aβs, and can further aggravate the cholinergic deficiency. M1 agonists can elevate APP, decrease tau protein phosphorylation/hyperphosphorylation, and restore cognitive impairments in several animal models for AD[32,33]. A significant decrease of M1 mAChR mRNA, but with no changes of M2, M3, or M4, in the temporal cortex of AD has been observed[34]. The M1 mAChR protein was decreased in the cortex and hippocampus despite unchanged levels of the M1 binding sites in the same regions[35]. The impaired neurotransmission of mAChRs might be involved in the pathogenesis of AD.
N-methyl-D-aspartate receptors in AD
Glutaminergic neurons form the major excitatory system in the brain and play an important role in brain functions. Glutamate activates several classes of metabotropic receptors, including 3 major types of inotropic receptors, such as N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-S-methylisoxazale-4-proprionic acid, and kainic acid receptors[36]. Essentially, the NMDA receptor has important functions in synaptic transmission, synaptic plasticity, synaptogenesis, and excitotoxicity[37].
There is growing evidence that disturbances of glutamin-ergic neurotransmission may contribute to the pathophysiological mechanism and cognitive deficits of AD. The colocalization of glutamatergic neurons and NFT or SP in the AD brain has been observed[38]. It has been reported that NMDA receptors are decreased in the cortical regions and hippocampus in AD brains[39]. A reduction in NMDA receptor subunit 1 mRNA in the hippocampus and an increase in the frontal and temporal cortices have also been observed[40]. In in vitro studies, the addition of Aβ enhances glutamate release from primary cultured microglia of rats[15] and increases the toxicity of glutamate[41]. The increase of vulnerability to glutamate-induced excitotoxicity has been found in the hippocampal neurons prepared from presenilin-1 mutation mice[42]. The studies by using transgenic mice with APP mutations have shown an enhanced sensitivity to glutamatergic agonists, such as NMDA and kainic acid[43]. Although almost all of the data have reported a decrease or no change of NMDA receptor subunits in AD, there is still ongoing debate as to whether NMDA receptor changes cause excitotoxicity or hypoactivity in AD[44]. This interest has been increased due to the drug approval of memantine, a partial NMDA antagonist, that can reduce clinical deterioration in moderate to severe AD[45].
Correlations between oxidative stress and modifications of cholinergic and glutaminergic receptors connected with AD
Both cholinergic and glutaminergic receptors are cellular membrane proteins that are located on the structure, which consists of membrane lipids. Studies on protein–lipid interactions have shown that functions of neurotransmitter receptors are highly dependent on the environment of lipid compositions[46]. Brain tissue is more vulnerable to free radical-mediated injury, because the organ utilizes high amounts of oxygen and its cellular membranes are preferentially enriched in oxyradical-sensitive polyunsaturated fatty acids[47]. Sequentially, the surrounding lipids of receptors in the brain are very sensitive to attack by free radicals. Oxidative stress has been implicated as a contributing factor to neurodegeneration in AD and has a significant connection with the deficits of cholinergic and glutaminergic receptors in the CNS.
Oxidative stress and nAChRs
Recently, we observed that lipid peroxidation in cellular membranes can induce a reduction in [3H]epibatidine or [125I]α-bungarotoxin (α-BTX) binding of nAChR in PC12 cells[18]. In addition, the free radical insult (FeSO4) we used, which it could induce concentration-dependent increases in lipid peroxidation, but did not result in apoptosis in PC12 cells, significantly induced reductions at the protein level for α3 and α7 nAChR subunits, and at the mRNA level for the α7 subunit[18], indicating an effect of free radicals on the gene expression of nAChR. The pretreatment of cultural cells with antioxidants, such as vitamin E and reduced glutathione, can prevent the inhibitory effect of free radicals on [3H]epibatidine and [125I]α-BTX-binding sites.
The correlation between oxidative stress and the loss of the α4 nAChR subunit has been investigated in the temporal cortex from patients with AD, and the results showed a significant correlation between increased levels of lipid peroxidation and decreased numbers of the α4 nAChR subunit protein in AD brains [17]. In a recent study, we also found that lipid peroxidation induced directly by Aβ might be involved in the deficits of nAChR[48]. In this study, PC12 cells were treated by the addition of 5 mmol/L Aβ25–35 and Aβ1–40, respectively, with or without an antioxidant, vitamin E. An increased lipid peroxidation and significant reductions in [3H]epibatidine and [125I]α-BTX binding sites and in the protein levels of the α3 and α7 nAChR subunits were observed in the cells treated with Aβ. Interestingly, the decreases of the nAChR binding sites and subunit proteins resulting from Aβ were mostly prevented by pretreatment with the antioxidant. Aβs damage and kill neurons possibly through an effect on membrane lipid peroxidation, impaired ion-motive ATPases, glucose, and glutamate transporters making nerve cells vulnerable to the excitotoxic effects of glutamate[49]. These findings suggest that lipid peroxidation induced by Aβ might trigger the loss of nAChR in AD[48].
Interestingly, the α7 nAChR subtype may have an antioxidative function. When being exposed to ethanol, PC12 cells exhibited an increase in intracellular oxidative stress, whereas after selectively activating α7 nAChR by a receptor agonist, 3-(2,4)-dimethoxybenzylidine anabaseine, the action of ethanol for stimulating oxidative stress can be attenuated[50]. These results suggest that the cytoprotection conferred by the α7 nAChR agonist may be mediated at least in part by reducing the formation or accumulation of ROS[50]. Recently, we observed that the decreased expression of α7 nAChR in SH-SY5Y cells by employing small interference RNA that are specifically targeted towards the receptor can enhance lipid peroxidation and stimulate the toxicity exerted by Aβ[51], suggesting that α7 nAChR plays a significant antioxidative role in connection with the pathogenesis of AD. The antioxidative properties connected with a mechanism through the activation of nAChR have also been demonstrated in PC12 cells by treatment with varying amounts of nicotine[52].
Oxidative stress and mAChR
Early studies have shown that oxidative stress can reduce the number of mAChR in the rat brain and canine heart[53]. The effects may be due to receptor destruction or inactivation resulting from membrane damage mediated by free radicals.
Recent findings have indicated that M1, M2, or M4 mAChR-transfected COS-7 cells show greater oxidative stress sensitivity than those transfected with M3 or M5, and similar findings have also been observed when the cells were exposed to Aβ25–35 and Aβ1–40, suggesting an attack on the receptors by free radicals induced by Aβ[54]. An endogenous inhibitor with low molecular weight from the AD brain inactivates the human brain mAChR via a decrease of ligand binding to the receptor[55]. The prevention of inactivation of the mAChR with antioxidants suggests that the endogenous inhibitor with low molecular weight generates damage of the receptor due to oxidative stress.
It has been demonstrated that ROS are generated after mAChR stimulation[56]. Blocking mAChR can prevent increased levels of oxidative stress. Since stimulation of the mAChR leads to the activation of multisignaling pathways, including the muscarinic-induced, mitogen-activated protein kinases pathways and ROS production, and to the upregulation of the binding activity of transcription factors, such as NF-κB and activator protein-1 (AP-1), ROS (over a narrow concentration range) might function as second messengers in cell-signaling pathways[57]. ROS may not merely cause damage, but may also be mediators of physiological functions.
Oxidative stress and NMDA receptors
It has been indicated that Aβ could place glutamate-sensitive neurons at risk by enhancing glutamate and oxygen free radical production by monocyte-derived cells, in which such mechanisms might contribute to the pathogenesis of AD. In glia cultures, Aβ inhibits glutamate uptake, probably connected with an increased production of free radicals[58]. Lipid peroxidation induced by homocysteine can be completely inhibited by the NMDA receptor antagonist, dizocilpine (MK-801), suggesting that oxidative injury to nerve terminals involves NMDA receptor stimulation[59]. Acute ammonia intoxication diminishes the activities of antioxidative enzymes and increases superoxide formation in rat brains, and the ammonia-induced oxidative stress is mediated by the excessive activation of NMDA receptors[60].
The protective action of stobadine, an antioxidant, can prevent the decreased number of NMDA binding sites elicited by hypoxia/reoxygenation in the rat hippocampus by its antioxidative and antiradical effects[61]. A low-affinity antagonist of NMDA receptors, sulfasalazine, can have neuroprotective effects by preventing Ca2+ influx and accumulation through blocking NMDA receptors[62]. Pyridostigmine bromide, a reversible cholinesterase inhibitor, can induce apoptosis in rat cerebellar granule cells by producing ROS generation, which can be blocked by MK-801 and atropine, a muscarinic receptor antagonist[63].
The mechanism of the influence of oxidative stress on membrane receptors might be involved in: (1) reactive oxygen metabolites affecting the binding of ligands to membrane receptors and also the coupling of receptors to G-proteins and effector enzymes; (2) the peroxidation of membrane lipids altering the viscosity of the plasma membrane, which affects receptor coupling; (3) ROS interaction with thiol/disulfide moieties on receptor proteins or on other factors in the receptor system, which is responsible for alterations in receptor binding or coupling; (4) the association of lipid peroxidation with the modification of fatty acids by the phospholipase A2 pathway, in which arachidonic acid binds directly to the receptors or perturbs the local environment to indirectly affect receptor function; (5) oxidative stress leading to a disturbance in cellular Ca2+ homeostasis, which might be related to an effect on Ca2+-mobilizing receptors; (6) ROS interference with actions of nitric oxide, thus affecting another pharmacological messenger system; and (7) oxidative stress influencing the processing of transcription or translation of these neurotransmitter receptors by modulating the expression at the protein or mRNA levels.
Conclusion
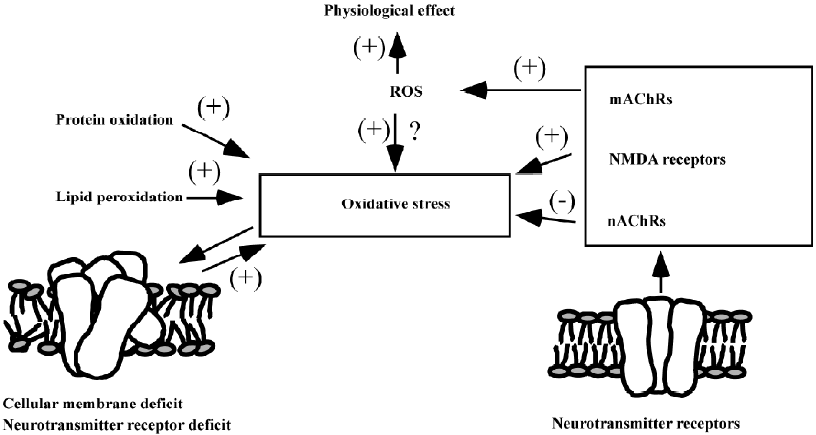
Both oxidative stress and the modifications of nAChR, mAChR, and NMDA receptors play important roles in the pathogenesis of AD. In addition, there is a closed relationship between oxidative stress and these receptors (Figure 3). A high level of free radicals can induce the disturbance of the cellular membrane by lipid peroxidation and attack protein receptors by protein oxidation, as well as possibly damaging the gene expression of these receptors by DNA oxidation, which might be an important mechanism for the receptor deficit in the AD process. nAChRs have also shown an antioxidative effect by a direct or indirect pathway, whereas high stimulation to NMDA receptors can increase the sensitivity of oxidative stress of neurons. mAChR stimulation can generate ROS, which might be a physiological compensative reaction, or improve oxidative stress. When considering the therapeutic strategy for AD, 2 more approaches with antioxidative properties and neurotransmitter receptor regulation might ultimately be combined to provide important and efficient roles in drug action.
References
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science 2002;298:789-91.
- Sorrentino G, Bonavita V. Neurodegeneration and Alzheimer’s disease: the lesson from tauopathies. Neurol Sci 2007;28:63-71.
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet 2006;368:387-403.
- Tariot PN, Federoff HJ. Current treatment for Alzheimer’s disease and future prospects. Alzheimer Dis Assoc Disord 2003;17:S105-113.
- Facchinetti F, Dawson VL, Dawson TM. Free radicals as mediators of neuronal injury. Cell Mol Neurobiol 1998;18:667-82.
- Betteridge DJ. What is oxidative stress? Metabolism 2000;49:3-8.
- Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med 2007;43:658-77.
- Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr 2000;71:S621-629.
- Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, et al. Oxidative damage in Alzheimer’s. Nature 1996;382:120-1.
- Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, et al. Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 1998;70:2212-5.
- Teunissen CE, de Vente J, Steinbusch HW, De Bruijn C. Biochemical markers related to Alzheimer’s dementia in serum and cerebrospinal fluid. Neurobiol Aging 2002;23:485-508.
- Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci 2001;21:4183-7.
- Pratico D. Alzheimer’s disease and oxygen radicals: new insights. Biochem Pharmacol 2002;63:563-7.
- Castellani RJ, Zhu X, Lee HG, Moreira PI, Perry G, Smith MA. Neuropathology and treatment of Alzheimer disease: did we lose the forest for the trees? Expert Rev Neurother 2007;7:473-85.
- Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta 2000;1502:139-44.
- Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer’s disease. J Neuropathol Exp Neurol 1999;58:740-7.
- Yu WF, Nordberg A, Ravid R, Guan ZZ. Correlation of oxidative stress and the loss of the nicotinic receptor alpha 4 subunit in the temporal cortex of patients with Alzheimer’s disease. Neurosci Lett 2003;338:13-6.
- Guan ZZ, Yu WF, Shan KR, Nordman T, Olsson J, Nordberg A. Loss of nicotinic receptors induced by beta-amyloid peptides in PC12 cells: possible mechanism involving lipid peroxidation. J Neurosci Res 2003;71:397-406.
- Smith MA, Casadesus G, Joseph JA, Perry G. Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med 2002;33:1194-9.
- Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol 2000;61:75-111.
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry 2001;49:200-10.
- Marutle A, Warpman U, Bogdanovic N, Lannfelt L, Nordberg A. Neuronal nicotinic receptor deficits in Alzheimer’s patients with the Swedish amyloid precursor protein 670/671 mutation. J Neurochem 1999;72:1161-9.
- Guan ZZ, Zhang X, Ravid R, Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J Neurochem 2000;74:237-43.
- Yu WF, Guan ZZ, Bogdanovic N, Nordberg A. High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp Neurol 2005;192:215-25.
- Guan ZZ, Miao H, Tian JY, Unger C, Nordberg A, Zhang X. Suppressed expression of nicotinic acetylcholine receptors by nanomolar beta-amyloid peptides in PC12 cells. J Neural Transm 2001.1417-33.
- Xiu X, Nordberg A, Shan KR, Yu WF, Thordman T, Olsson JM, et al. Lovastatin stimulates up-regulation of alpha7 nicotinic receptors in cultured neurons without cholesterol dependency, a mechanism involving production of the alpha-form of secreted amyloid precursor protein. J Neurosci Res 2005;82:531-41.
- Dineley KT, Bell KA, Bui D, Sweatt JD. Beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 2002;277:25056-61.
- Bednar I, Paterson D, Marutle A, Pham TM, Svedberg M, Hellstrom-Lindahl E, et al. Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol Cell Neurosci 2002;20:354-65.
- van Koppen CJ, Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther 2003;98:197-220.
- Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA 1996;93:13541-6.
- Svensson AL, Warpman U, Hellstrom-Lindahl E, Bogdanovic N, Lannfelt L, Nordberg A. Nicotinic receptors, muscarinic receptors and choline acetyltransferase activity in the temporal cortex of Alzheimer patients with differing apolipoprotein E genotypes. Neurosci Lett 1997;232:37-40.
- Gu Z, Zhong P, Yan Z. Activation of muscarinic receptors inhibits beta-amyloid peptide-induced signaling in cortical slices. J Biol Chem 2003; 278: 17 546–56.
- Genis I, Fisher A, Michaelson DM. Site-specific dephosphorylation of tau of apolipoprotein E-deficient and control mice by M1 muscarinic agonist treatment. J Neurochem 1999;72:206-13.
- Wang SZ, Zhu SZ, Mash DC, el-Fakahany EE. Comparison of the concentration of messenger RNA encoding four muscarinic receptor subtypes in control and Alzheimer brains. Brain Res Mol Brain Res 1992;16:64-70.
- Flynn DD, Ferrari-DiLeo G, Mash DC, Levey AI. Differential regulation of molecular subtypes of muscarinic receptors in Alzheimer’s disease. J Neurochem 1995;64:1888-91.
- Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: preclinical evidence. Int J Geriatr Psychiatr 2003;18:S23-32.
- Dunah AW, Yasuda RP, Luo J, Wang Y, Prybylowski KL, Wolfe BB. Biochemical studies of the structure and function of the N-methyl-D-aspartate subtype of glutamate receptors. Mol Neurobiol 1999;19:151-79.
- Francis PT, Sims NR, Procter AW, Bowen DM. Cortical pyramidal neuron loss may cause glutamatergic hypoactivity and cognitive impairment in Alzheimer’s disease: investigative and therapeutic perspectives. J Neurochem 1993;60:1589-604.
- Greenamyre JT, Penney JB, Young AB, D’Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science 1985;227:1496-9.
- Panegyres PK, Zafiris-Toufexis K, Kakulas BA. The mRNA of the NR1 subtype of glutamate receptor in Alzheimer’s disease. J Neural Transm 2002;109:77-89.
- Brorson JR, Bindokas VP, Iwama T, Marcuccilli CJ, Chisholm JC, Miller RJ. The Ca2+ influx induced by beta-amyloid peptide 25-35 in cultured hippocampal neurons results from network excitation. J Neurobiol 1995;26:325-38.
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 1999;5:101-6.
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 1999;274:6483-92.
- Lee HG, Zhu X, Ghanbari HA, Ogawa O, Raina AK, O’Neill MJ, et al. Differential regulation of glutamate receptors in Alzheimer’s disease. Neurosignals 2002;11:282-92.
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003;348:1333-41.
- Fong TM, McNamee MG. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry 1986;25:830-40.
- Choe M, Jackson C, Yu BP. Lipid peroxidation contributes to age-related membrane rigidity. Free Radic Biol Med 1995;18:977-84.
- Guan ZZ, Zhang X, Mousavi M, Tian JY, Unger C, Nordberg A. Reduced expression of neuronal nicotinic acetylcholine receptors during the early stages of damage by oxidative stress in PC12 cells. J Neurosci Res 2001;66:551-8.
- Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, et al. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol 1998;44:819-24.
- Li Y, King MA, Meyer EM. alpha7 nicotinic receptor-mediated protection against ethanol-induced oxidative stress and cytotoxicity in PC12 cells. Brain Res 2000;861:165-7.
- Qi XL, Nordberg A, Xiu J, Guan ZZ. The consequences of reducing expression of the α7 nicotinic receptor by RNA interference and of stimulating its activity with an α7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer’s disease. Neurochem Int 2007;51:377-83.
- Guan ZZ, Yu WF, Nordberg A. Dual effects of nicotine on oxidative stress and neuroprotection in PC12 cells. Neurochem Int 2003;43:243-9.
- Arora RC, Hess ML. Effect of reduced oxygen intermediates on sarcolemmal muscarinic receptors from canine heart. Biochem Biophys Res Commun 1985;130:133-40.
- Joseph JA, Fisher DR. Muscarinic receptor subtype determines vulnerability to amyloid beta toxicity in transfected cos-7 cells. J Alzheimers Dis 2003;5:197-208.
- Fawcett JR, Bordayo EZ, Jackson K, Liu H, Peterson J, Svitak A, et al. Inactivation of the human brain muscarinic acetylcholine receptor by oxidative damage catalyzed by a low molecular weight endogenous inhibitor from Alzheimer’s brain is prevented by pyrophosphate analogs, bioflavonoids and other antioxidants. Brain Res 2002;950:10-20.
- Mangelus M, Kroyter A, Galron R, Sokolovsky M. Reactive oxygen species regulate signaling pathways induced by M1 muscarinic receptors in PC12M1 cells. J Neurochem 2001;76:1701-11.
- Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal 1999;11:1-14.
- Harris ME, Wang Y, Pedigo NW Jr, Hensley K, Butterfield DA, et al. Amyloid beta peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 1996;67:277-86.
- Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A. Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res 2003;5:237-43.
- Kosenko E, Kaminski Y, Lopata O, Muravyov N, Felipo V. Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Radic Biol Med 1999;26:1369-74.
- Gasparova-Kvaltinova Z, Stolc S. Effect of antioxidants and NMDA antagonists on the density of NMDA binding sites in rat hippocampal slices exposed to hypoxia/reoxygenation. Methods Find Exp Clin Pharmacol 2003;25:17-25.
- Ryu BR, Lee YA, Won SJ, Noh JH, Chang SY, Chung JM, et al. The novel neuroprotective action of sulfasalazine through blockade of NMDA receptors. J Pharmacol Exp Ther 2003;305:48-56.
- Li L, Shou Y, Borowitz JL, Isom GE. Reactive oxygen species mediate pyridostigmine-induced neuronal apoptosis: involvement of muscarinic and NMDA receptors. Toxicol Appl Pharmacol 2001;177:17-25.