
P53-mediated cell cycle arrest and apoptosis through a caspase-3-independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells
Introduction
Herbal medicine, Donglingcao (rabdosia rubescens), has been traditionally used in China for the treatment of leukemia. Oridonin is a diterpenoid compound isolated from rabdosia rubescens. It has potent antitumor functions[1] and has been used for the treatment of human cancers, especially esophageal carcinoma[2]. This compound has been observed to prevent mutation, decrease Na+-pump transportation activity of cancer cells, and promote the efficiency of other antitumor agents[3-5]. It induces apoptosis to inhibit cancerous cell proliferation, as well as enhances the sensitivity of drug-resistant cancer cell lines[6,7].
Apoptosis is an essential and highly conserved mode of cell death that is important for normal development, host defense, and the suppression of oncogenesis. Apoptosis removes cancerous or virally-infected cells, and aberrant apoptosis is the major cause for tumor development and progression[8]. Among the numerous proteins and genes involved, members of caspase family and the Bcl-2 family play pivotal roles in modulating apoptosis. Apoptosis is mediated by the activation of caspases, which amplify the apoptotic signal and proteolytically process numerous cellular target molecules with various functions[9]. Bcl-2 family proteins are vital for the regulation of apoptosis by controlling the mitochondrial membrane potential to release cytochrome c and for activating caspase-9 and -3[10,11]. The balance between pro-apoptotic proteins and anti-apoptotic proteins determines the fate of cells.
The DNA damage response, caused by a variety of stimuli, arrests the cell cycle to allow damage repair or direct cell apoptosis[12]. An imbalance between DNA damage and DNA repair activities may affect cell viability. After DNA damage, the cell cycle is arrested at the transition from the G1 to S phase or from the G2 to M phase of the cell cycle. Increasing evidence indicates a central role for p53 in mediating cell cycle arrest or apoptosis[13,14].
In the present study, we demonstrated that activated p53 contributed to oridonin-induced cell cycle arrest and apoptosis, and mitochondrial alternations amplified the activation of the caspase cascade; meanwhile, caspase-9, together with calpain rather than caspase-3, led to oridonin-induced apoptosis in human breast MCF-7 cells.
Materials and methods
Chemicals Oridonin was obtained from Kunming Institute of Botany (The Chinese Academy of Sciences, Kunming, China), and the structure of oridonin was assigned by comparing the chemical and spectral data (1H-NMR, 13C-HMR) with those reported in other published literature[15]. The purity of oridonin was measured by HPLC and determined to be more than 99%. Oridonin was dissolved in dimethyl sulfoxide (DMSO) to make a stock solution. DMSO concentrations were kept at below 0.05% in all the cell cultures and did not exert any detectable changes in cell growth or apoptosis. Fetal bovine serum (FBS) was purchased from TBD Biotechnology Development (Tianjin, China); propidium iodide (PI), Rhodamine 123, and RNase A were purchased from Sigma Chemical (St Louis, MO, USA). Thiazolyl blue (MTT) was from Sino-American Biotechnology (Beijing, China); Z-VAD-fmk and calpain inhibitor II were bought from Sigma Chemical (St Louis, MO, USA); rabbit polyclonal antibodies against Bax, p53, p-p53, p21, caspase-9 and -3, PARP, heat shock protein (Hsp)90, the inhibitor of caspase-activated DNase (ICAD), mouse polyclonal antibodies against Bcl-2, and horseradish peroxidase-conjugated secondary antibody (goat-anti-rabbit or goat-anti-mouse) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture The MCF-7 cell line (#CRL HTB-22) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in RPMI-1640 medium (GIBCO,LA, NY, USA) supplemented with 10% FBS and 0.03% L-glutamine (GIBCO, USA) and maintained at 37 °C with 5% CO2 in a humidified atmosphere.
Cell viability assay The cytotoxic effect of oridonin on the MCF-7 cells was measured by MTT assay as described in previous studies[16]. The cells were dispensed in 96-well, flat bottom microtiter plates (NUNC, Roskilde, Denmark) at a density of 1?104 cells per well. After 24 h incubation, they were treated with various concentrations of oridonin, followed by 12, 24, 36, and 48 h cell culture. 20 µL MTT solution (5.0?103 mg/L) was added to each well 4 h before the end of incubation. The effects of Z-VAD-fmk and calpain inhibitor II on oridonin treated MCF-7 cells were also determined by MTT assay, and both inhibitors were added into the cell culture 1 h before oridonin administration, respectively. The resulting crystals were dissolved in DMSO. Absorbance was measured with an ELISA reader (TECAN SPECTRA, Wetzlar, Germany). The cytotoxic effect was expressed as a relative percentage of inhibition calculated as follows:
Relative inhibition (%)=[(A 490 control - A 490 oridonin)/A 490 control] ×100
Observation of morphological changes The MCF-7 cells were plated in the wells of a 6-well plate at a density of 3×105 cells per well. After cultured for 24 h, the cells were treated with 80 µmol/L oridonin and incubated for 12 or 24 h. The cellular morphological changes were observed using phase contrast microscopy (Leica, Nussloch, Germany).
Membrane leakage assay The integrity of the plasma membrane of the MCF-7 cells was determined by monitoring the cell lactate dehydrogenase (LDH) leakage, which was accomplished by following the rate of conversion of NADH to NAD+. LDH released into the extracellular medium was expressed as a percentage of total LDH[17,18]. In brief, 1.5×106 cells were cultured with oridonin for 0, 8, 16, 24, and 36 h. Floating dead cells were collected from the culture medium by centrifugation, and the LDH content from the pellets lysed in 0.1% NP-40 for 15 min was used as an index of apoptotic cell death (LDHp). The LDH released in the culture medium [extracellular LDH (LDHe)] was used as an index of necrotic cell death. The LDH released from adherent and viable cells was expressed as intracellular LDH (LDHi). The substrate reaction buffer of LDH was added. The A value at 490 nm of reaction for 1 and 5 min was assayed. LDH activity=(A5 min-A1 min)/4. The percentage of apoptotic and necrotic cell death was calculated as follows:
% apoptosis=[LDHp/(LDHp+LDHe+LDHi)] ×100
% necrosis=[LDHe/(LDHp+LDHe+LDHi)] ×100
Mitochondrial transmembrane potential alternation Mitochondrial transmembrane potential (Δψmit) alternation was determined by Rhodamine 123 staining. For the fluorescence observation, the MCF-7 cells were seeded into 6-well plates and mounted on the coverslips. For the flow cytometric analysis, the cells were harvested by tripsin. After culture with oridonin for 12 h, the cells were removed from the culture medium for staining. The culture medium was replaced with phosphate buffered solution (PBS) and washed 3 times. The cells were incubated in Rhodamine 123 staining stock solution (5 g/L) for 20?30 min at 37 °C. The coverslips were washed 3 times in PBS and mounted with the stained cells for fluorescent microscopy. Mitochondrial transmembrane potential changes were indirectly determined by measuring Rhodamine 123 fluorescence variance using a cytoflowmeter with an emission wavelength of 525 nm and an excitation wavelength of 488 nm. The samples were quantified as quickly as possible[19-21].
DNA extraction and detection of DNA fragmentation The MCF-7 cells (2×106 cells) were harvested with pancreatin and centrifuged at 1000×g for 10 min. The cell pellets were suspended in 10 mmol Tris-HCl (pH 7.4), 10 mmol edetic acid 0.5% Triton X-100, and 40 µg/L proteinase K (Merck, WS, NJ, USA) at 37 °C for 2 h. The lysate was extracted with 0.5% 5 mol/L NaCl and 50% 2-propanol and incubated overnight at -20 °C, and then centrifuged at 7 000×g for 20 min. The supernatant was washed with 70% ethanol and centrifuged at 7 000×g and the pellets were dried and resuspended in10 mmol/L Tris-HCl (pH 7.4) and 1 mmol/L edetic acid. DNA was incubated with 40 µg/L RNase A (Sigma, St Louis, MO,USA) at 37 °C for 60 min, separated by 2% agarose gel electrophoresis at 100 V for 40 min, and stained with 0.1 mg/L ethidium bromide[22,23].
Flow cytometric analysis After the oridonin treatment, the sample preparation was performed as previously described[24,25]. 1×106 MCF-7 cells were harvested and washed once in cold PBS. The cell pellets were fixed in 75% ethanol at 4 °C overnight and washed in cold PBS. Then the pellets were suspended in 1 mL of 50 mg/L PI solution, 0.1% (w/v) sodium citrate, and 0.1% (v/v) Triton X-100. The cell samples were incubated at 4 °C in the dark for at least 15 min, and analyzed by a FACScan flowcytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blotting 2×106 MCF-7 cells were pre-incubated with or without 2 mmol/L 3-methyladenine (3-MA) for 1, then treated with 64 μmol/L oridonin for 12, 24, 36, or 48 h. Both adherent and floating cells were collected and Western blotting was performed as previously described[26]. Briefly, the MCF-7 cells were washed with ice-cold PBS and solubilized with lysis buffer (1% SDS, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L EDTA, 2 mmol/L leupeptin, and 1 mmol/L aprotinin). The protein concentration was determined by the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). The protein lysates were separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were soaked in blocking buffer (5% skimmed milk in PBS), and then incubated overnight with primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies. The color was developed with diaminobenzidine (DAB).
Statistical analysis All data represent at least 3 independent experiments and are expressed as the mean±SD unless otherwise indicated. Statistical comparisons were made by Students’s t-test. P-values of less than 0.05 were considered significant.
Results
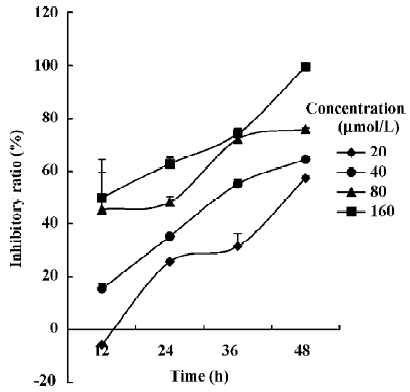
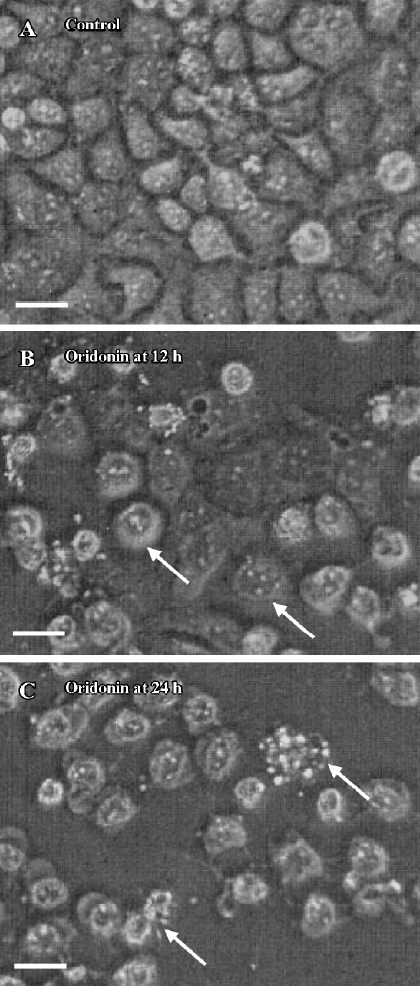
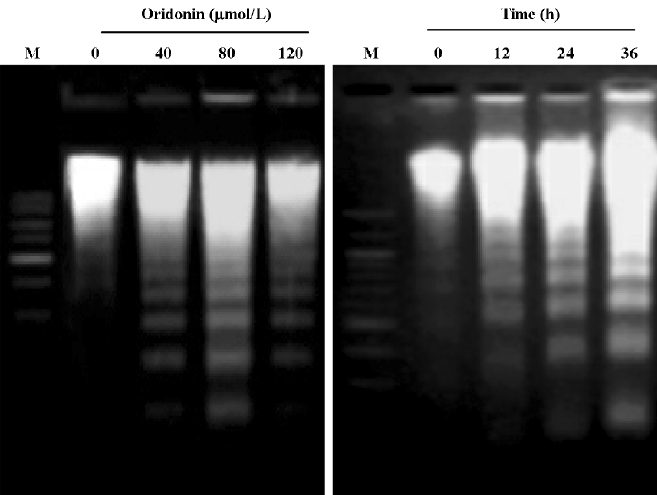
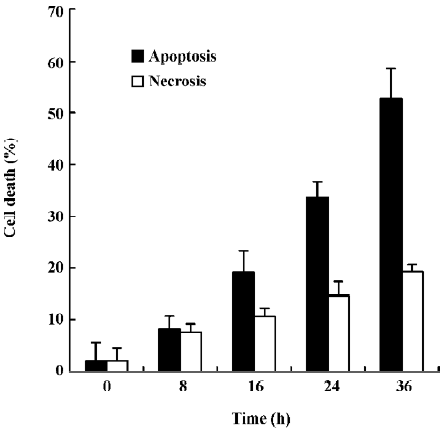
Oridonin induced apoptosis in MCF-7 cells Oridonin induced MCF-7 cell death in a time- and concentration-dependent manner. The IC50 value of 24 h of oridonin treatment for the MCF-7 cells was 84.18 µmol/L. The treatment of the MCF-7 cells with 80 µmol/L oridonin for 24 h induced approximately 48.3% of the growth inhibition (Figure 1). Mean-while, marked morphological changes were observed compared with the untreated control. The oridonin-treated MCF-7 cells underwent retraction of cellular processes and became round in shape at 12 h (Figure 2B). By 24 h, the majority of the MCF-7 cells had shrunk in shape. Blebbing nuclei and granular apoptotic bodies appeared, and some cells were almost floating (Figure 2C). Untreated cells did not show these apoptotic characteristics (Figure 2A). After treatment with 80 µmol/L oridonin for 12, 24, and 36, h with 40, 80, and 120 µmol/L oridonin respectively the MCF-7 cells began to generate typical DNA fragmentation, which is a hallmark of apoptosis (Figure 4). The number of apoptotic cells increased from 8.24% at 8 h to 52.74% at 36 h in the presence of oridonin, while the necrotic cell proportion also increased from 7.58% at 8 h to 19.24% at 36 h (Figure 3). However, the percentage of necrotic cells was still negligible compared with that of apoptotic cells. These results demonstrated that treatment with oridonin induced the majority of MCF-7 cell apoptosis.
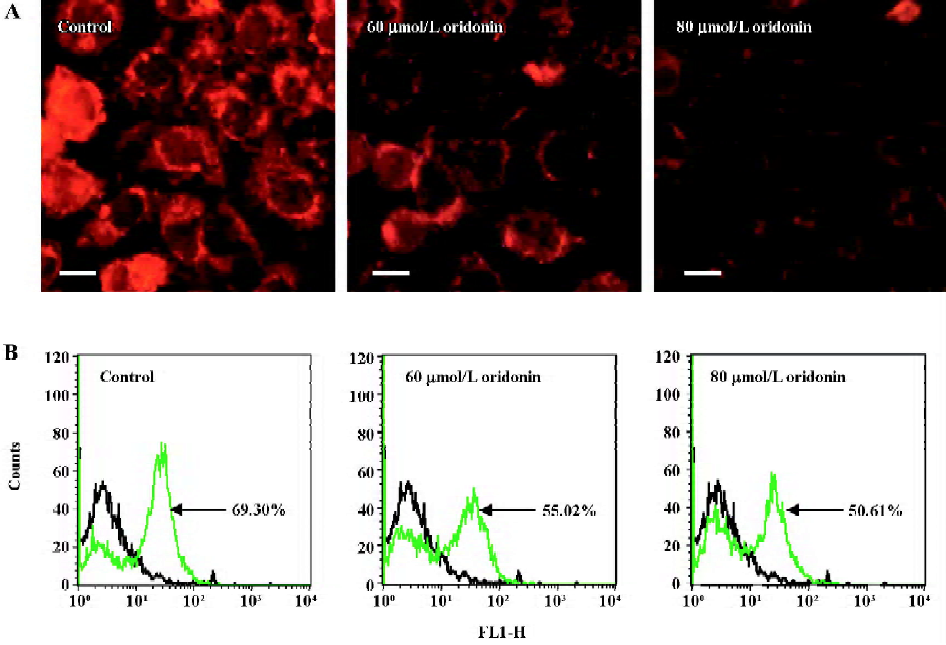
Oridonin induced disruption of mitochondrial integrity Rhodamine 123 was first used to measure the Δψmit in intact cells, both as a microscopic stain and by cytoflowmeter by monitoring the increase in fluorescence due to its electrophoretic accumulation in mitochondria. In isolated mitochondria, energization induced a red shift and extensive quenching of Rhodamine 123 fluorescence; therefore, dye accumulation could be suggested as a sensitive and specifically fluorescent potentiometric probe of Δψmit of mitochondria in living cells[27]. The oridonin-treated MCF-7 cells displayed a specifically lower Rhodamine fluorescent density (Figure 5A) and lower transmembrane potential (Figure 5B) compared with the untreated cells, indicating that oridonin damaged the mitochondrial respiratory chain and triggered apoptosis. Moreover, the alternations of fluorescence and transmembrane potential were both in a dose-dependent manner in oridonin-challenged MCF-7 cells.
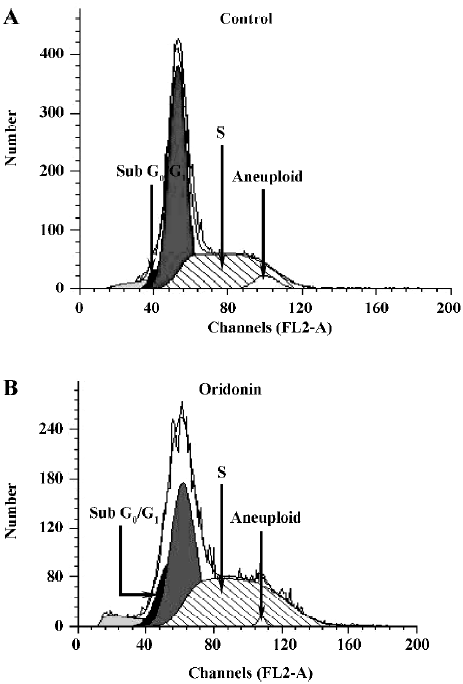
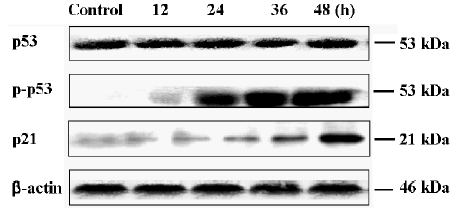
Oridonin induced S phase arrest and the upregulation of p53 and p21 To investigate further features of cell growth inhibition by oridonin, a flow cytometric analysis was performed. After treatment with oridonin for 12 h, the cells were accumulated in the S phase. The Sub-G0/G1 peak denoting apoptosis was clearly observed (Figure 6). Simul-taneously, aneuploid-presented tumor cells were declined after oridonin treatment (Table 1). Meanwhile, phosphorylated p53 and p21 proteins were also both upregulated at 12 h, while non-phosphorylated p53 remained the same (Figure 7).
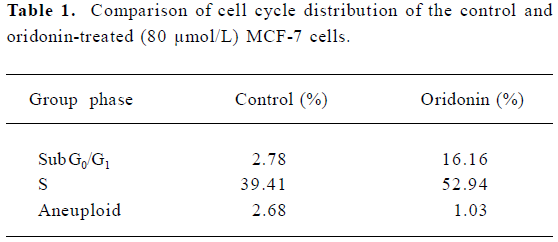
Full table
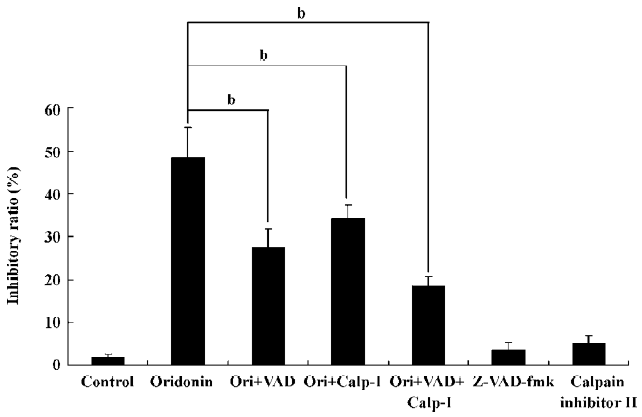
Both caspases and calpain facilitated cell death Caspases are unique cysteine proteases that are synthesized as inactive precursors and are activated during apoptosis. Among them, caspase-3 is a common downstream apoptosis effector, which is processed and activated by caspase-9 or -8 and digested into the heterodimeric form (17-12 kDa) by mitochondrial pathways or through the activation of death-domain containing receptors[28]. Calpain, like caspase-3, is a cytosolic cysteine protease, but requires Ca2+ for its activity. Ubiquitous calpain exists in resting cells, but it is activated by Ca2+ and triggers autolytic processing; it is also activated in some apoptosis systems[29]. To investigate the influences of caspases and calpain on apoptosis, we applied their inhibitors into experiment systems. 10 µmol/L pan-caspase inhibitor Z-VAD-fmk and/or 20 µmol/L calpain inhibitor II was introduced into the cell culture, then after 1 h, 80 µmol/L oridonin was added. Both inhibitors exerted protective effects on cell growth, indicating that caspases and calpain accelerated oridonin-induced MCF-7 cell death. Thus, we further investigated whether the combined use of these two inhibitors in the cells could be sufficient to block the cell death. The resulting data showed that the inhibitory ratio further declined, compared with single inhibitor administration of each inhibitor, but was not as low as the control level, indicating that there might be some other factor(s) influencing cell death (Figure 8).
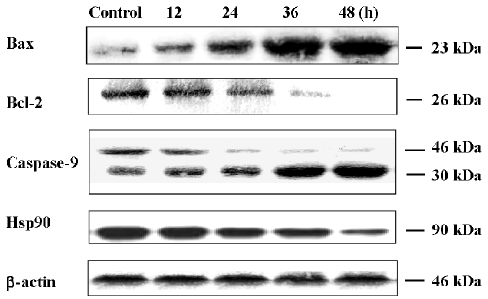
Effects of oridonin on the expression of Bax, Bcl-2, caspase-9, and Hsp90 Since anti- and pro-apoptotic members of the Bcl-2 family arbitrate the survival-or-death decision, we detected the expressions of Bcl-2 and Bax by Western blot analysis. After the oridonin treatment, the expression of Bcl-2 decreased; on the contrary, the expression level of Bax increased in a time-dependent manner. Caspase-9 was a pivotally effective apoptotic protease in the postmitochondrial pathway. In the present study, under the condition of oridonin employment, procaspase-9 was cleaved, and activated caspase-9 was enhanced in a time-dependent manner. Hsp90 displayed protective functions in many cell lines[14]. Here, when the MCF-7 cells were exposed to oridonin, the Hsp90 expression was significantly downregulated (Figure 9).
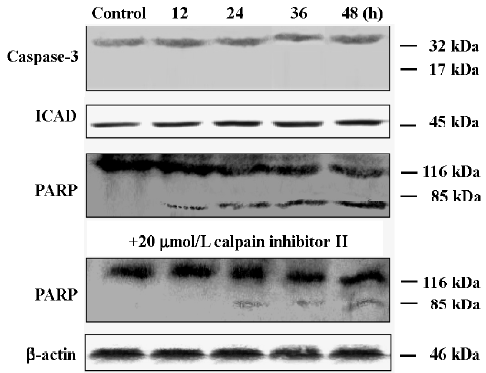
Expressions of caspase-3, PARP, and ICAD ICAD was a classical substrate of caspase-3. After the oridonin treatment, the expression of ICAD was unchanged (Figure 10), which seemed to be consistent with the viewpoint that apoptosis in the MCF-7 cells was not through caspase-3[30]. To further confirm this hypothesis, we detected the expressions of caspase-3 and PARP. In the MCF-7 cells, procaspase-3 was not cleaved into the activated form. However, PARP was activated and formed the 85 kDa isoform which was digested from its 116 kDa precursor in a time-dependent manner, indicating that other enzymes activated PARP. According to this speculation, we examined a more promising candidate, calpain, since it was reported that PARP was a calpain substrate[36]. However, unexpectedly, under calpain inhibitor II application, the cleavage of the PARP precursor was partially blocked. 116 kDa PARP precursors were almost not discerned disparity, and 85 kDa PARP protein expressions decreased, suggesting that calpain partially participated in PARP activation, inducing apoptosis, and bypassing caspase-3.
Discussion
In this study, we have demonstrated that oridonin inhibited cell growth, arrested the cell cycle, and induced apoptosis in MCF-7 cells.
These biochemical events were possibly associated with the p53 tumor suppressor gene. The p53 protein displayed a key role of p53 in the G1/S checkpoint in response to DNA damage[13] as a regulator of cell cycle progression and a mediator of apoptosis in many cell lines. In response to various types of DNA damage, the cell cycle checkpoints and cell death signals are activated to stop cell growth and to eliminate multiplication of the genetically-altered cells. Damaged cells stop DNA replication at the G1 or G2 phase, presumably allowing the repair systems to function before the next cell cycle. Apoptosis is also triggered in response to various DNA damage. The activation of the apoptotic cell death pathway is a safeguard in removing irrepairably damaged cells. Several cellular effector molecules, including p53, are involved in arresting damaged cells at these checkpoints and inducing apoptosis. The upregulation of the p53 protein is a common cellular response in many cell types expos-ed to various DNA damaging agents[14,31].
The cell cycle progression was decelerated by p53 and cdk inhibitors, including p21[32]. The cyclin dependent kinase inhibitor, p21, is a multifunctional protein involved in coordinating the cellular response to negative growth signals. Induced by cellular damage under the transcriptional control of the p53 tumor suppressor protein, p21 interfaces with a number of cellular proteins involved in growth control. p21 plays an essential role in growth arrest after DNA damage, and their overexpression leads to G1 and G2 phase arrest[33]. Therefore, the activation of this signaling pathway has been considered to be important for the efficacy of antitumor agents, and direct transactivation of the p21 gene by passing p53 can serve as a novel strategy for treating cancers that are insensitive to classical antitumor agents. Our results show that oridonin caused cell cycle arrest in the S phase through the upregulation of p53 and p21. Here, activated p53, after DNA damage by oridonin, might either trigger the onset of DNA repair, leading to the completion of the cell cycle, or lead to the exit from the cell cycle, apoptosis via mitochondria.
Hsps are a large family of highly-conserved proteins broadly categorized according to their size and functions, which expressed under stress, usually confer survival protection to the cell or interruption in the apoptotic pathways. Among the Hsps, the Hsp90 family is ubiquitously expressed, and is one of most abundant cytoplasm proteins. Hsp90 can physically interact with either the mutant or the wild type p53 in vivo[34], to partially block the apoptotic progression. On the other hand, Hsp90 forms a cytosolic complex with Apaf-1 and thereby inhibits the formation of the active Apaf-1– caspase-9 apoptosome complex to negatively control apoptosis[35]. Our findings demonstrate that Hsp90 declined with oridonin application, and further confirmed that p53 exerted a double function on both cell cycle arrest and apoptosis in oridonin-treated MCF-7 cells. On one hand, p53 through p21 provoked cell cycle arrest at the S phase to repair DNA damage by oridonin treatment in the MCF-7 cells; on the other hand, p53 promoted apoptosis via Bax/Bcl-2 to clean irrepairable cells from cell cycle arrest, which might be an explanation as to why the oridonin-treated MCF-7 cells did not arrest the cell cycle and apoptosis at the same time in the present experiment system.
Caspases play a crucial role in the apoptotic progression, morphological changes, and DNA fragmentation suggesting that oridonin-induced MCF-7 cell death was involved in a mechanism of apoptosis. One of the major pathways for caspase activation involves the participation of mitochondria. Bcl-2 inhibits the apoptotic process and promotes cell survival, and Bax acts in the mitochondria to cause the release of cytochrome c, leading to the activation of caspase-9, and the subsequent activation of caspase-3[36]. Moreover, Bax expression is regulated by p53 and the protein products of the target genes of p53, including Bcl-2, are involved in this process[37]. In the present study, oridonin decreased the Bcl-2 expression and activated Bax, suggesting that the mitochondria was involved in oridonin-induced apoptosis.
Upon cytochrome c release into the cytoplasm, Apaf-1 is activated and triggers the caspase cascade. One of the key events in this pathway is the caspase-3-mediated cleavage of the ICAD, which allows caspase-activated DNase to enter the nucleus and causes oligonucleosomal DNA fragmen-tation. In this study, ICAD was left unaffected due to no active form of caspase-3 rather than lack of capase-3 expression, which was different from the view that the MCF-7 cell line was deficient in procaspase-3 according to previous studies[38], but DNA fragmentation was induced in this study. More recently, it has been discovered that in response to apoptotic stimuli, mitochondria can also release caspase-independent cell death effectors such as apoptosis inducing factor (AIF). AIF could induce nuclear apoptosis in a variety of cell types and this effect was not inhibited by pharmacological caspase inhibitors such as zVAD, indicating that AIF can trigger nuclear apoptosis in a caspase-independent manner. AIF binds to DNA in a sequence-independent manner, which determines the entry of this complex into the nucleus. It recruits or activates an endonuclease to facilitate DNA fragmentation and chromatin condensation[39]. Thus, it was probable that DNA was fragmented by AIF in our experiment system. Meanwhile, capase-9 was upregulat-ed; we could speculated that after the mitochondrial and postmitochondrial caspase-9-dependent pathways were activated, other effective caspases such as caspases-6 or -7, but not caspase-3, might take responsibility for apoptotic signal transduction. In addition, it was worth noting the participation of calpain, a Ca2+-dependent intracellular cysteine protease in oridonin-treated MCF-7 cell death. Calpain is activated in various necrotic and apoptotic conditions, while caspase-3 is only activated in apoptosis. Caspases and calpains share several substrates, including PARP, and during apoptosis, the 116 kDa PARP is degraded by caspase-3 to distinct 89 and 27 kDa fragments; however, recently it has been found to be cleaved by calpain at alternative sites, generating fragments from 70 to 40 kDa in size during necrosis[40]. Furthermore, growing evidence suggests that calpain may play a central role in the execution of apoptosis. Our results show that calpain inhibitor II could partially rescue oridonin-induced MCF-7 cell death and PARP cleavage, indicating that calpain contributes to cell death and replaces caspase-3 to execute PARP activation in part. All data here allowed us to speculate that other enzymes existed to induce the apoptotic signals. Caspase-7 might substitute for capase-3 in most cell types and tissues[41], since it is highly homologous to caspase-3 and has very similar substrate specificity. Further studies determining the factor(s) prohibiting caspase-3 cleavage remain to be conducted.
Maintenance of a significant electrical potential difference across biological membranes is crucial for a variety of cellular functions, including development, signaling, movement, energy balance, and apoptosis. Intracellular organelles such as mitochondria possess function-related membrane potentials far exceeding that of the plasma membrane. The dissipation of the inner mitochondrial transmembrane potential marks the point of no return during the apoptotic program and occurs prior to DNA fragmentation. Thus, the evaluation of mitochondrial transmembrane potential depolarization is of critical importance for the assessment of apoptosis[42,43]. As our results demonstrated, exposure to oridonin caused an emission of Rhodamine fluorescence which represented the declined mitochondrial membrane potential, inferring that oridonin enhanced apoptosis concomitantly with a decrease in Δψmit.
In summary, oridonin inhibited MCF-7 cell growth and arrested cell cycle through the activation of p53 in response to DNA damage, and decreased cdk activities by p21 was involved in the apoptotic progression. Simultaneously, oridonin induced cell apoptosis mediated by p53, through the upregulation of Bax and the downregulation of Bcl-2 and Hsp90, which contributed to the activation of caspase-9, leading to the activation of downstream caspases in the process. Moreover, calpain bypassed caspase-3 to partially contribute to oridonin-induced MCF-7 cell death.
References
- Kitamura K, Okuda Y, Ikekawa N, Kada T, Nomoto M. Anti-mutagenic diterpenoids from a crude drug (Enmei-so). Agric Biol Chem 1984; 48: 1647-8.
- Zhang TM. Recent studies on the antitumor activity of rabdosia rubescences. Chin J Oncol 1982;4:322-3. Chinese..
- Lin BL, Yang DP, Hu HY. Molecular pharmacology and clinic application strategy in anti-cancer of oridonin. Guangdong Pharm J 2000;10:5-8. Chinese..
- Yang MH, Jiang B, Zhao QS, Sun HD. Studies on diterpenoids of isodon sculponeata. Chin Tradit Herb Drugs 2001;32:397-9. Chinese..
- Yang SL, Han SY, Zhang Q, Song AY, Ding LP, Gong YO, et al. Study on the antimutagenicity of oridonin. Carcin Terat Mut 2001;13:8-9. Chinese..
- Liu JY, Pan XL, Wu XY, Cai GQ. The mechanism of apoptosis induced by oridonin in K562 cells. Cancer Res Clin 2000;24:47-8. Chinese..
- Guo JJ, Pan XL, Feng CW, Zou JH. Study on reversal effects of oridonin on multidrug resistant cell line K562/A02. Shanghai Med J 2002;25:43-5. Chinese..
- Han DKM, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G. Evidence for apoptosis in human atherogenesis and in a rat vascular injury modal. Am J Pathol 1995;147:267-77.
- Cohen GM. Caspase: the executioners of apoptosis. Biochem J 1997;326:1-16.
- Akgul C, Moulding DA, Edwards SW. Molecular control of neutrophil apoptosis. FEBS Lett 2001;487:318-22.
- Zhi YH, Panayotis P, James HW, Nichola K, Vincent JK, Eric AH. A Fas-associated death domain protein-dependent mechanism mediates the apoptotic action of non-steroidal anti-inflammatory drugs in the human leukemic Jurkat cell line. J Biol Chem 2001;276:38748-54.
- Lozano G, Elledge SJ. p53 sends nucleotides to repair DNA. Nature 2000;404:24-5.
- Ciciarello M, Mangiacasale R, Casenghi M, Limongi MZ. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem 2001;276:19205-13.
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282:1497-501.
- Zhang CL, Wu LJ, Tashiro S, Onodera S, Ikejima T. Oridonin induces apoptosis of HeLa cells via altering expression of Bcl-2/Bax and activating caspase-3/ICAD pathway. Acta Phamacol Sin 2004;25:691-8.
- Lasek W, Wankowicz A, Kuc K, Feleszko W, Golab J, Giermasz A, et al. Potentiation of antitumor effects of tumor necrosis factor alpha and interferon gamma by macrophage-colony-stimulating factor in a MmB16 melanoma model in mice. Cancer Immunol Immunother 1995;40:315-21.
- Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases on caspase-3-like activity via two distinct mechanisms. J Biol Chem 1997;272:31138-47.
- Zhang Y, Wu LJ, Tashiro S, Onodera S, Ikejima T. Evodiamine induces tumor cell death through different pathways: apoptosis and necrosis. Acta Pharmacol Sin 2004;25:83-9.
- Ma J, Cui FZ. Measuring membrane potential and electric field of brainstem neurons in vitro by confocal microscopy. Brain Res Brain Res Protoc 2004;13:84-90.
- Baracca A, Sgarbi G, Solaini G, Lenaz G. Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F0 during ATP synthesis Biochim Biophys Acta 2003;1606:137-46.
- Zhu YG, Chen XC, Chen ZZ, Zeng YQ, Shi GB, Su YH, . Curcumin protects mitochondria from oxidative damage and attenuates apoptosis in cortical neurons. Acta Pharmacol Sin 2004; 25: 1606-12.
- Sarin A, Hadid E, Henkart PA. Caspase dependence of target cell damage induced by cytotoxic T lymphocytes. J Immunol 1998;161:2810-3.
- Gong YG, Chen DD, He GJ, Guan YY. Effect of 15-deoxy-delta12,14-postaglandin J2 on cell proliferation and apoptosis in ECV304 endothelial cells. Acta Pharmacol Sin 2004;25:47-53.
- Lee KY, Park JA, Chung E, Lee YH, Kim SI, Lee SK. Ginsenoside Rh2 blocks the cell cycle of SK-KEP-1cells at the G1/S boundary by selectively inducing the protein expression of p27kipl. Cancer Lett 1996;110:193-200.
- Yang SR, Wen L, Lu YQ, Gong QY, Yu R, Yao MH. Effects of GM-CSF, IL-3 and GM-CSF/IL-3 fusion protein on apoptosis of human myeloid leukemic cell line Tf-1 induced by irradiation. Acta Pharmacol Sin 2004;25:68-75.
- Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, et al. Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1 beta. J Immunol 2001;167:5940-7.
- Baracca A, Sgarbi G, Solaini G, Lenaz G. Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F0 during ATP synthesis. Biochim Biophys Acta 2003;1606:137-46.
- Nicholson DW, Thornberry NA. Caspases, killer proteases. Trends Biochem Sci 1997;22:299-306.
- Nakagawa T, Yuan JY. Cross-talk between two cysteine protease families activation of caspase-12 by calpain in apoptosis. J Cell Biol 2000;150:887-94.
- Liang Y, Nylander KD, Yan C, Schor NF. Role of caspase 3-dependent Bcl-2 cleavage in potentiation of apoptosis by Bcl-2. Mol Pharmacol 2002;61:142-9.
- Smith ML, Fornace AJ Jr. Mammalian DNA damage-inducible genes associated with growth arrest and apoptosis. Mutat Res 1996;340:109-24.
- Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 1993;8:307-18.
- Xu ZM, Wang YQ, Mei Q, Chen J, Du J, Wei Y, et al. Effects of p21 (Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998;18:629-43.
- Zhang Y, Wang JS, Chen LL, Zhang Y, Cheng XK, Heng FY, et al. Repression of hsp90beta Gene by p53 in UV irradiation-induced apoptosis of Jurkat cells. J Biol Chem 2004;279:42545-51.
- Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J 2000;19:4310-22.
- Colman MS, Afshari CA, Barrett JC. Regulation of p53 stability and activity in response to genotoxic stress. Mutat Res 2000;462:179-88.
- Kurland JF, Meyn RE. Protease inhibitors restore radiation induced apoptosis to Bcl-2-expressing lymphoma cells. Int J Cancer 2001;96:327-33.
- Kagawa S, Gu J, Honda T, McDonnell TJ, Swisher SG, Roth JA, et al. Deficiency of caspase-3 in MCF7 cells blocks bax-mediated nuclear fragmentation but not cell death. Clin Cancer Res 2001;7:1474-80.
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004;23:2785-96.
- McGinnis KM, Gnegy ME, Park YH, Mukerjee N, Wang KKW. Procaspase-3 and poly (ADP) ribose polymerase (PARP) are calpain substrates. Biochem Biophys Res Commun 1999;263:94-9.
- Wang KKW. Calpain and caspase: can you tell the difference? Trends Neurosci 2000;23:20-6.
- Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, . Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death . J Exp Med 1995; 181: 1661-72.
- Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med 1995;182:367-77.