
Detection and analysis of bovine foamy virus infection by an indicator cell line1
Introduction
Foamy virus (FV) belongs to the retroviridae family and can infect a wide range of hosts, including chimpanzees, monkeys, cats, and cows[1,2]. The virus undergoes reverse transcription (RT) in the cytoplasm and integrates into the host genome upon viral infection. Like most retroviruses, the genomic structure of FV from 5' to 3' includes 5' long terminal repeat (LTR), gag, pol, env, and 3' LTR[1]. However, FV presents some unique features that are distinct from other members of retroviridae[3]. A few regulatory genes, bel-1 (Tas), bel-2, bel-3, and bet are located between env and 3' LTR[4,5]. An internal promoter and a cis-acting element[6,7] were identified within env gene sequence and a region from 5' LTR to pol, respectively. In addition, the replication strategy and viral genomic contents of FV are also quite different from most retroviruses, but present marked similarities with hepadnaviruses[3].
Traditional titration of the infectious FV is usually achieved by using the cytopathic effect (CPE)[8]. However, not all the cells that are infected by FV show CPE[9,10]. Therefore, the method based on CPE may have a limitation on the titration of FV. Other methods, such as RT-PCR, which has been reported for the detection of prototype foamy viruses[11,12], does not give the direct quantitation on viral loading during infection. Recently, a new method called the green fluorescent protein (GFP)-based indicator cell line was established for the titration of prototype foamy virus (PFV) and feline foamy virus (FeFV)[13,14]. The indicator cell line was constructed by the stable integration of the GFP gene under the control of the foamy virus LTR promoter. Viral titration can be monitored by GFP expression after FV infection. This method is rapid and accurate in detecting FV infection and is very useful for clinical diagnosis as well as basic research.
It has long been noted that both RNA and infectious DNA genomes are present in extracellular FV virions[15,16]. Azidothymidine (AZT), a potent inhibitor of reverse transcri-ptase, has been introduced to the indicator cell line of FV for the detection of the time point of RT during PFV and FeFV infections[3]. Studies indicate that the RT of FV is not at the early phase of infection, but occurs late in the replication cycle before new virus buds from an infected cell. This is held true for both human and feline FV[16–18]. However, the step of RT in bovine foamy virus (BFV) has not been defined.
In this study, we employed the indicator cell line system to assay the infectivity of BFV in 3 different cells in a non-pathogenic condition. In addition, the replication strategy of BFV was evaluated by assaying the effect of AZT on BFV production.
Materials and methods
Cell culture Fetal bovine lung (FBL) cells, 293T, HeLa, and the BFV indicator cell line (BICL) cells were preserved by our laboratory (Laboratory of Molecular Virology, Nankai University, Tianjin, China). The BICL, 293T, and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 105 U/L penicillin, and 0.1 g/L streptomycin. The FBL cells were cultured in DMEM supplemented with 10% FCS, 105 U/L penicillin, 0.1 g/L streptomycin, and non-essential amino acids. All the cells were cultured at 37 °C in 5% CO2.
Reagents AZT and Lipofectamine was purchased from Invitrogen (Carlsbad, CA, USA). DMEM was purchased from Gibco (Carlsbad, CA, USA). The pre-stained molecular weight markers were purchased from Bio-Red (Hercules, CA, USA). Other chemicals used in the Western blotting and in the β-galactosidase enzyme assay in this study were of analytical grade and commercially available.
Preparation of virus The strain BFV3026 was isolated from peripheral lymphoid cells by our laboratory[19]. The virus was harvested and treated in 3 different ways: (i) after being infected with BFV3026, the infected cells were trypsinized and gathered for the co-culture experiment; (ii) after being infected with BFV and centrifuged at 4 000×g for 10 min, the cell supernatant was filtered through a 0.22 µm filter membrane for subsequent use; and (iii) after transfection of FBL, 293T, and HeLa cells with the full-length BFV proviral clone pCMV (cytomegalovirus) - BFV, the cells were trypsinized and gathered for the co-culture experiment.
Plasmid construction pcDNA3.1(-) was purchased from Invitrogen (USA). To generate the infectious BFV molecular clone pCMV-BFV, we assembled the subgenomic BFV clones step by step based on the provirus clone pBS (short form of pbluescript)-BFV. The U3 promoter region of 5' LTR was replaced by the immediate early gene promoter of human CMV in pBS-BFV to generate pCMV-BFV.
DNA transfections and β-galactosidase enzyme assay The cells were plated in 6-well plates (2×105/well) and cultured at 37 °C for 24 h. After that, the cells were transfected with 3 μg pCMV-BFV plasmid and 0.5μg pSV-β-galactosidase control vector (Promega, Madison, WI,, USA) to each well according to the manufacturer’s instructions (N
GFP activation assay by BFV The BICL cells were plated at a density of 2×105 per well in 6-well plates in DMEM medium. On the following day, the medium was removed and the cells were incubated with infected cells through the co-culture method or incubated with the supernatant of infected cells. Forty-eight hours after co-culturing, the infected cells were observed and counted for GFP expression under an inverted fluorescence microscope (Olympus IX-71, Shinjuku-ku, Tokyo, Japan). Subsequently, the total BICL cells were trypsinized and counted with a hemocyto-meter. The percentage of GFP-positive cells was calculated by the ratio of GFP positive cells against the total cultured cells.
SDS-PAGE and Western immunoblotting The infected cells were mixed with 5×sample buffer containing 0.6 mol/L Tris-HCl (pH 6.8), 25% glycerol, 2% SDS, and 0.1% bromophenol blue, and incubated for 5 min at 100 °C. 50 µL of each sample was loaded and electrophoresed for 80 min at the constant voltage of 100 V. After electrophoresis, polypeptides and markers separated on each gel were electrotrans-ferred onto a 0.2 µm nitrocellulose membrane overnight at 30 V under chilled conditions. Each membrane was blocked with a 1% skim milk solution by submerging the membrane in the solution for 30 min at a room temperature.
For the Western immunoblot analysis, the nitrocellulose membrane was incubated with an antibody against BTas (Tas of BFV) at a dilution of 1:5000 in 20 mmol/L Tris-buffered saline solution with 0.05% Tween 20 (TBST, pH 7.5) for 90 min at an ambient temperature with slow rocking, and then rinsed at least 3 times with TBST. Then the membrane was incubated with goat anti-mouse IgG or IgM labeled with horseradish peroxidase (Santa Cruz biotechnology, Santa Cruz, CA, USA) for 60 min at an ambient temperature. After the developing and fixing process, the membrane was exposed to Kodak XAR-5 film (KODAK (CHINA) LIMITED, Beijing, China). β-actin was detected as a loading control; the β-actin antibody was purchased from Sigma (St Louis, MO, USA).
The result was photographed by AlphaImager 2200 (Alpha Innotech, San Leandro, CA, USA) and analyzed by ImageJ software (Research Services Branch, National Institute of Mental Health, Bethesda, Maryland, USA). The integrated density of each band represented the protein expression levels.
AZT inhibition experiments The cell lysate prepared from BFV-infected FBL cells were used to infect 2×105 FBL, 293T, and HeLa cells through the co-culture method at a ratio of 1:10. The infected cells were co-cultured with the indicator cells at 48 h post infection. Then the BICL cells were tested by FV-activated GFP assay. The final concentration of AZT was 25 µmol/L.
Results
Evaluation of BFV infectivity in non-cytopathic conditions by BICL BFV preferentially propagates in bovine fibroblastic cells such as FBL. We found that no significant CPE was induced in human cell lines such as HeLa and 293T cells after incubation with excessive BFV3026. However, the infectivity of BFV on these cells was unclear.
In order to clarify this issue, a BICL system was used for the detection of viral infectivity. This system is very sensitive since the infectivity of BFV can be detected by monitoring GFP expression even in a non-cytopathic state. The infectivity of BFV against FBL, HeLa, and 293T cells was evaluated by an indicator cell line through a co-culture approach. We infected the above cell lines at low MOI (multiplicity of infection) to ensure that all cells infected did not induce CPE within 48 h post infection. Then the infected cells were co-cultured with 2×105 BICL cells at a ratio of 1:10 (Table 1). The activation of the GFP expression could be induced in BICL after being co-cultured with the BFV-infected cells (Figure 1). The GFP-positive cells were counted. Here we defined the GFP-positive percentage as 1. The ratio of these 3 cell lines was 1.00: 0.19: 0.17.

Full table
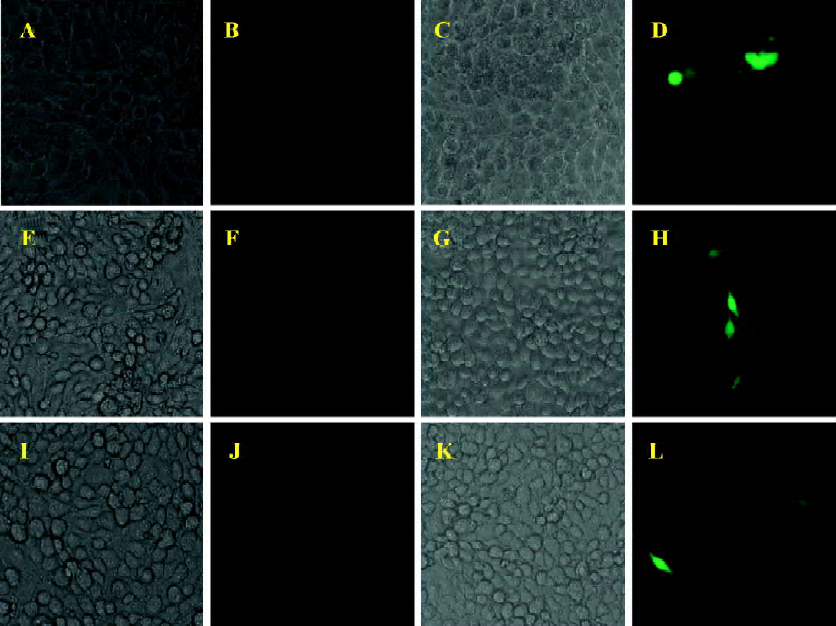
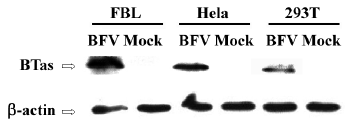
Moreover, we detected the BFV infection on the expression level. FBL, HeLa, and 293T cells were infected at the same MOI. Forty-eight hours after the infection, these infected cells were lysed, and the expression of BTas was detected by Western immunoblotting (Figure 2). The integrative density ratio of BFV-infected FBL, HeLa, and 293T cells was 1.00:0.22:0.15 (Table 2).

Full table
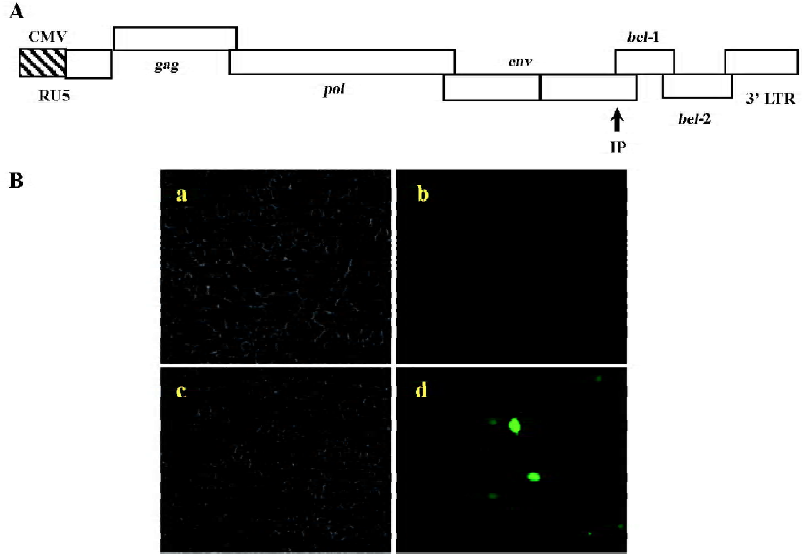
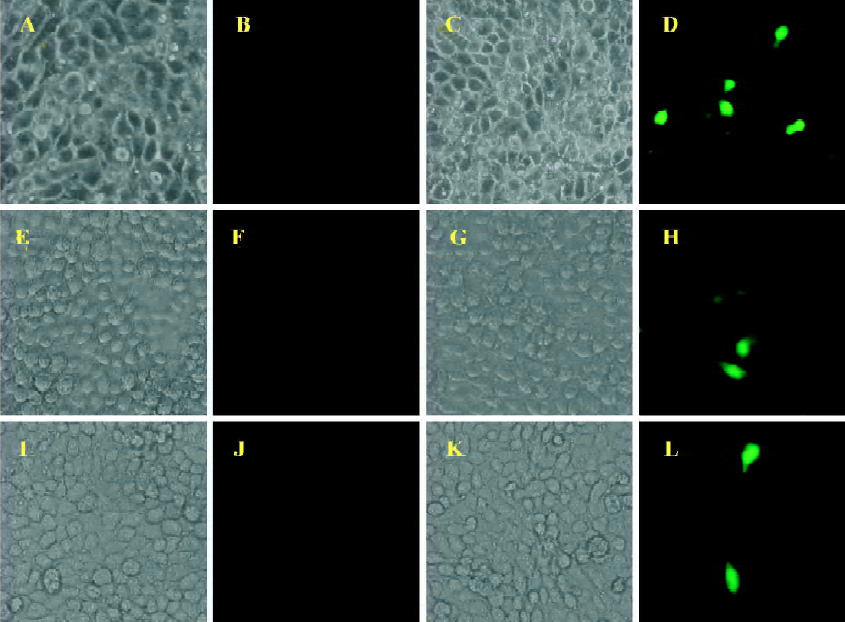
Using BICL to detect the infectivity of plasmid pCMV-BFV in mammalian cells pCMV-BFV was the infectious clone of BFV driven by the human CMV immediate early gene promoter instead of its U3 promoter region of 5' LTR (Figure 3A). FBL cells were first transfected with pCMV-BFV and incubated for 4 d. The transfected FBL cells were co-cultured with BICL cells at a ratio of 1:10. The induction of the GFP expression could be clearly observed 2 d later (Figure 3B), indicating that pCMV-BFV can successfully generate infectious BFV viruses in the cells. Similarly, in non-cytopathic cell lines such as HeLa and 293T, we also detected the GFP expression (Figure 4). However, the infectivity produced by pCMV-BFV in HeLa and 293T cells (1.62%±0.40% and 1.33%±0.47%, respectively) was lower than that in FBL cells (2.52%±0.78%; Table 3). Their initial was 1.00: 0.53: 0.64. After transfection efficiency, the β-gal report system was employed to calibrate this result. The ratio after calibration was 1.00: 0.21: 0.14 (Table 3).
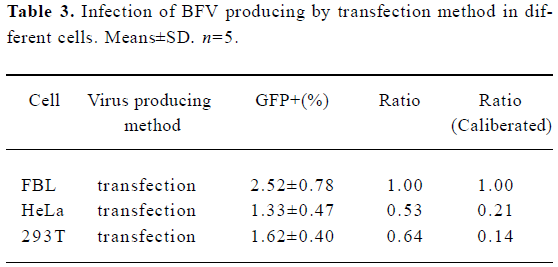
Full table
Inhibitory effect of AZT on BFV replication FBL, 293T, and HeLa cells were first infected with BFV3026 at a ratio of 1:10. Then these cells were either treated with 25 µmol/L AZT or left untreated for 48 h. The titers of BFV3026 in these cells were determined on BICL indicator cells which had been pretreated with AZT for 24 h or left untreated. When the virus was produced and assayed in the absence of AZT, the virus replicated very well in all 3 cells and led to high titers of infectious virus 48 h after infection (Table 4), positive control, experiment I). When AZT was present during virus production and in the BICL indicator cells, almost few virus replications were detected (Table 4), negative control, experiment II), indicating that AZT effectively inhibited the RT. BFV titers decreased when AZT was absent from virus production and present in the viral titration assay (experiment III in Table 4). Remarkably, the viral titer was down to the negative control level when AZT was absent from the production of BFV and present during the titration assay (Table 4, experiment IV).
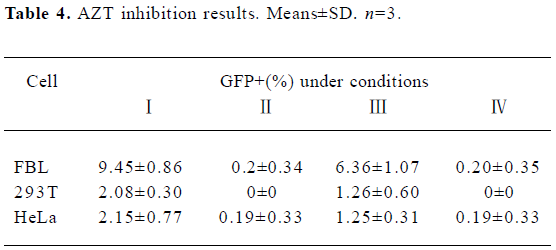
Full table
Discussion
Quantitative detection of viral infection is important for biological studies of FV infection. In this study, we demonstrate that the titration of BFV can be achieved by using a GFP-based indicator cell line system. This system is very useful and sensitive in the detection of BFV replication in non-cytopathic conditions. In addition, we have successfully used this indicator cell line system to show that the RT of BFV also occurs at the late phase during BFV infection.
Although FV can strongly induce CPE in a broad range of infected cells in vitro, the viruses usually do not cause clinical syndromes in natural hosts[2]. FV often cause apathogenic or persistent FV infection in certain cell types such as primary monocyte, leukemia, and epithelial cancer cell lines[14,20,21]. Thus, traditional CPE-based FV titration is greatly restricted in these non-cytopathic cells. As an sensitive assay, RT-PCR has been used for the identification of FV infection in cultured cells, experimental animals, or natural hosts[12,17,22]. However, because of the hard-to-handle standard test, it is difficult for RT-PCR to quantitate viral infection, thus it may not be used widely for viral titration. In 1993, Yu et al constructed an indicator cell line system and used this system to detect prototype FV infection by employing beta-galactosidase gene as an indicator[23]. Later studies demonstrated that other reporter genes, such as luciferase, could also be used for HFV titration[12,24]. However, both systems need either fixation with a special chemical substrate or the destruction of infected cells by lysis buffer. Therefore, it could not be directly applied to viral detection in living cells. Recently, a GFP-based indicator cell assay has been introduced for the titer detection of FeFV infection, and has proved to be a convenient and sensitive approach[9].
In agreement with these findings, our result provides further evidence to show that the GFP-based indicator cell line system is indeed sensitive and can be applied for the detection of BFV infections, even at non-cytopathic conditions. Our results show that in the co-culture experi-ment, the percentage of GFP-positive BICL infected by FBL-BFV was about 5fold more than that of HeLa-BFV and 293T-BFV. Western immunoblotting result also show different expression levels of BTas, and the ratio among these 3 cells shows the same trend as that of BICL detection. However, in the transfection experiment, the pCMV-BFV-transfected FBL cells were only about 1.5-fold higher than that of HeLa and 293T in the BICL system. This discrepancy is likely due to the low transfection efficiency of plasmid DNA as compared with the high efficiency of viral genome delivery during viral infection. As the β-gal report result shows, the transfection efficiency of FBL cells is lower than those of HeLa and 293T cells. Moreover, the calibrated ratio correlates well with that of BFV infection experiment. As a result, in both experiments, the infectivity detected in FBL cell was significantly higher than that in human cells such as 293T and HeLa cells, indicating that the propagation of BFV in these human cells was limited. This restriction was suspected to be because of the defects in viral entry, viral replication, the aberrant expression of either Tas or Bet[25], or epigenetic modification of BFV proviral genomes[9].
Using a potent RT inhibitor AZT, Rethwilm and colleagues restricted HFV and FeFV at a late stage of virus infection[17,18]. Similarly, we employed the GFP gene as an indicator to replace the beta-galactosidase gene in the AZT assay, which allowed us to observe viral infection in a living cell system. Our results are consistent with their studies and indicate that RT was a necessary and essential step for BFV production that took place before the virus release from an infected cell.
References
- Delelis O, Lehmann-Che J, Saib A. Foamy viruses–a world apart. Curr Opin Microbiol 2004;7:400-6.
- Falcone V, Schweizer M, Neumann-Haefelin D. Replication of primate foamy viruses in natural and experimental hosts. Curr Top Microbiol Immunol 2003;277:161-80.
- Lochelt M. Foamy virus transactivation and gene expression. Curr Top Microbiol Immunol 2003;277:27-61.
- Flugel RM, Rethwilm A, Maurer B, Darai G. Nucleotide sequence analysis of the env gene and its flanking regions of the human spumaretrovirus reveals two novel genes. EMBO J 1987;6:2077-84.
- Lochelt M, Muranyi W, Flugel RM. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc Natl Acad Sci USA 1993;90:7317-21.
- Wu M, Chari S, Yanchis T, Mergia A. cis-Acting sequences required for simian foamy virus type 1 vectors. J Virol 1998;72:3451-4.
- Heinkelein M, Schmidt M, Fischer N, Moebes A, Lindemann D, Enssle J, et al. Characterization of a cis-acting sequence in the Pol region required to transfer human foamy virus vectors. J Virol 1998;72:6307-14.
- Parks WP, Todaro GJ. Biological properties of syncytium-forming (“foamy”) viruses. Virology 1972;47:673-83.
- Schweizer M, Fleps U, Jackle A, Renne R, Turek R, Neumann-Haefelin D. Simian foamy virus type 3 (SFV-3) in latently infected Vero cells: reactivation by demethylation of proviral DNA. Virology 1993;192:663-6.
- Miyazawa T, Itagaki S, Tomonaga K, Ikeda Y, Mori T, Kawaguchi Y, et al. Establishment of carrier-state infection of a feline renal cell line with feline syncytial virus. J Vet Med Sci 1995;57:65-9.
- Lagaye S, Vexiau P, Morozov V, Guenebaut-Claudet V, Tobaly-Tapiero J, Canivet M, . Human spumaretrovirus-related sequences in the DNA of leukocytes from patients with Graves disease. Proc Natl Acad Sci USA 1992; 89: 10 070–4.
- Sun KH, Lin HY, Chen LW, Tai HY, Lin ML, Feng CK, et al. Human foamy virus bel1 sequence in patients with autoimmune rheumatic diseases. Clin Rheumatol 2005.1-6.
- Li Z, Yang P, Liu H, Li WX. A new indicator cell line to monitor human foamy virus infection and stability in vitro. Intervirology 2002;45:79-84.
- Phung HT, Tohya Y, Shimojima M, Kato K, Miyazawa T, Akashi H. Establishment of a GFP-based indicator cell line to quantitate feline foamy virus. J Virol Methods 2003;109:125-31.
- Yu SF, Sullivan MD, Linial ML. Evidence that the human foamy virus genome is DNA. J Virol 1999;73:1565-72.
- Delelis O, Saib A, Sonigo P. Biphasic DNA synthesis in spumaviruses. J Virol 2003;77:8141-6.
- Roy J, Rudolph W, Juretzek T, Gartner K, Bock M, Herchenroder O, . Feline foamy virus genome and replication strategy. J Virol 2003; 77: 11 324–31.
- Moebes A, Enssle J, Bieniasz PD, Heinkelein M, Lindemann D, Bock M, et al. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J Virol 1997;71:7305-11.
- Liu SH, Chen HX, Chen JT, Liang D, Chen QM, Geng YQ, et al. Isolation and identification of a bovine spuma virus isolate 3026. Virol Sin 1997;13:140-5. Chinese..
- Gould EA, Hartley J. Comparison of the antigens produced by foamy virus in a cytolytic and a persistent infection of HEp2 cells. J Gen Virol 1979;44:235-9.
- Meiering CD, Rubio C, May C, Linial ML. Cell-type-specific regulation of the two foamy virus promoters. J Virol 2001;75:6547-57.
- Mikovits JA, Hoffman PM, Rethwilm A, Ruscetti FW. In vitro infection of primary and retrovirus-infected human leukocytes by human foamy virus. J Virol 1996;70:2774-80.
- Yu SF, Linial ML. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J Virol 1993;67:6618-24.
- Tai HY, Sun KH, Kung SH, Liu WT. A quantitative assay for measuring human foamy virus using an established indicator cell line. J Virol Methods 2001;94:155-162.
- Meiering CD, Linial ML. Reactivation of a complex retrovirus is controlled by a molecular switch and is inhibited by a viral protein. Proc Natl Acad Sci USA 2002;99:15130-5.