
Verapamil blocks HERG channel by the helix residue Y652 and F656 in the S6 transmembrane domain1
Introduction
The human-ether-a-go-go-related gene (HERG)[1] encodes the pore-forming α-subunits of channels that conduct the rapid delayed rectifier K+ current IKr[2]. IKr is one of the most important membrane currents responsible for ventricular action potential repolarization. The suppression of HERG channels can lead to electrocardiographic changes including action potential and QT interval prolongation, which can be both antiarrhythmic and cause long-QT syndrome (LQTS)[3]. More commonly, LQTS is an adverse effect of many different types of drugs, including antiarrhythmics, antihistamines, antibiotics, gastrointestinal prokinetics, and antipsychotics[4,5]. It has been documented that drug-induced QT prolongation is mainly due to the drug-mediated inhibition of IKr, although these drugs are structurally diverse[4–7].
Structure function data provides evidence that the HERG channel possesses a larger pore cavity than other voltage-gated K+ (Kv) channels. Moreover, the aromatic amino acids which present in the inner (S6) helices of HERG are absent from Kv channels, which form key components of a high-affinity drug binding site. These features appear to confer upon the HERG channel a unique susceptibility to pharmacological blockade[8].
Because of their potential pro-arrhythmic effects, a number of non-cardiac drugs have been withdrawn from the market (eg terfenadine, cisapride, sertindole, grepafloxacin, and thioridazine) and many have been labeled for restricted use (eg mesoridazine, ziprasidone, droperidol, astemizol, and arsenic trioxide). Therefore, screening compounds for HERG and QT interval liability is now routine in the pharmaceutical industry. To facilitate the rational design of safer drugs without HERG liability, it is important to understand the biophysical and molecular mechanisms of HERG blocked by drugs. Verapamil, an L-type calcium antagonist, is useful in the treatment of hypertension, stable angina, and narrow QRS complex supraventricular arrhythmias. In addition to blocking L-type Ca2+ channels, verapamil is a potent antagonist of IK[9–11]. In a previous study, it was reported that wild-type (WT) HERG is expressed in human embryonic kidney (HEK 293) cells with an estimated half-maximal inhibition concentration (IC50) of 143.0 nmol/L, and the C-type inactivation-deficient mutations, Ser620Thr and Ser631Ala, lied in the S5-S6 linker near the internal and the external mouth of the channel pore, reduced verapamil blockade, which is consistent with a role for C-type inactivation in high-affinity drug blockade[10]. At the same time, Lang and colleagues showed that verapamil and its enantiomers inhibited HERG K+ channels expressed in Xenopus oocytes with half-maximal inhibitory concentration (IC50) values of 2.2–4.0 µmol/L[11]. However, until now, the biophysical and molecular mechanisms of HERG blockade by verapamil have been unclear. Accordingly, we used HERG expressed in oocytes to investigate the inhibitory action on HERG K+ channel current (IHERG) by verapamil, and determined whether or not key molecular determinants of HERG blockade for previously investigated drugs[12] are also important for the inhibition of IHERG by verapamil.
Materials and methods
Oocyte preparation Oocytes were isolated by dissection from adult Xenopus laevis. The frogs were anaesthetized by ice for 20–30 min. After dissection and the removal of the ovarian lobes, the incision was sutured and the frogs were allowed to recover for about 1 month before the removal of a second set of oocytes. Clusters of oocytes were digested with 1.5 mg/mL Type IA collagenase (Sigma, St Louis, MO, USA) in a Ca2+-free ND96 solution which contained 96 mmol/L NaCl, 2 mmol/L KCl, 2 mmol/L MgCl2, and 5 mmol/L N-2-hydroxy-ethylpiperazine-N'-2-ethanesulfonic acid (HEPES), pH 7.6 for 1.5 h, and washed extensively with Ca2+-free solution without collagenase.
cRNA preparation and injection HERG channel site-directed mutagenesis (Y652A and F656A) were chosen for the study (Figure 1). Wild-type Y652A and F656A HERG cDNA were generously donated by Dr Michael SANGUINETTI (Utah University, Salt Lake City, Utah, USA). Complementary RNA for injection into oocytes were prepared with the mMESSAGE mMACHINE kit (Ambion, Austin, TX, USA) after linearization of the expression construct with EcoR I.
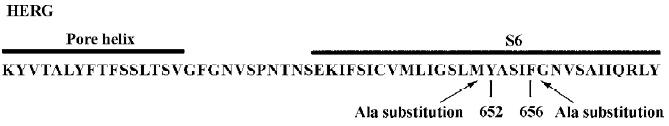
Stage V–VI deflocculated X laevis oocytes were injected with 50 nL cRNA (1 µg/µL) per oocyte using a Nanoject microdispenser (Drummond Scientific, Broomhall, PA, USA) and incubated at 18 °C in ND96 solution (96 mmol/L NaCl, 2 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, and 5 mmol/L HEPES, pH 7.6) supplemented with 100 U/mL penicillin.
Electrophysiological experiments Electrophysiological measurements were performed 2–10 d after oocyte injection. The 2-microelectrode voltage clamp was used to record currents from X laevis oocytes. The microelectrodes were filled with 3 mol/L KCl and had a resistance of 1–5 MΩ. Recordings were performed using a commercially available amplifier (Warner OC-725A, Warner Instruments, Hamden, CT, USA) and pCLAMP software (Axon Instruments, Foster City, CA, USA) for data acquisition and analyses. Currents were recorded in an ND 96 bath solution at room temperature (20–23 °C). The current signals were low-pass-filtered at 500 Hz and no leak subtraction was used. Oocytes were kept in the current-clamp mode for at least 5 min before switching to voltage-clamp mode. Only oocytes exhibiting a resting potential less than -40 mV were used.
Drugs Verapamil (Sigma, USA) was prepared as a stock solution (25 mmol/L). Before the experiments, the stock solution was diluted with bath solution to reach the desired final concentration. During the experiments, the cell-superfusate was exchanged using a home-built solution application device capable of changing the solution bathing a cell in less than 10 s. In general, recordings were started 60 s after the solution switch. All measurements were performed under steady-state conditions at least 2 min after the total solution exchange.
Statistical analysis The digitized data were analyzed with pCLAMP software (Axon Instruments, USA) and ORIGIN software (Origin Lab Corporation, Northampton, MA, USA) and Excel (Microsoft, Redmond, WA, USA). The concentration-response curves were fitted with a logistic dose-response equation to obtain the IC50 values. To determine the voltage dependence of HERG current activation, a least squares algorithm on ORIGIN software or Excel was used to fit tail current amplitudes (Itail) to a Boltzmann equation. The corrected steady-state inactivation curves were also fitted with a Boltzmann equation. All values are presented as mean±SEM. An analysis by Student’s t-test was performed for paired or unpaired observations. A P value of less than 0.05 was considered significant.
Results
Voltage-dependent block of WT and Y652A HERG channels by verapamil The voltage dependence of IHERG blockade was determined using the protocol shown in the inset of Figure 2A. Representative WT and Y652A HERG current traces in the control and in the presence of verapamil are respectively shown in Figure 2A, 2B. Both activating currents measured at the end of the depolarizing step (Istep) and peak tail current amplitude (Itail) measured following the step to -60 mV were dramatically reduced by verapamil (Figure 2). After the application of 10 µmol/L verapamil, the peak of the I-V relationship for both channels were shifted to the left (Figure 2C, 2D), suggesting a negative shift in the voltage dependence of activation. This was confirmed by a tail current analysis (Figure 2G, 2H). The current-voltage plot for tail currents (control Itail and verapamil Itail at -60 mV) was fitted with a Boltzmann equation to obtain the mid-point activation voltage (V1/2) and slope factor. For WT HERG channels, the half-point activation value was -19.73±0.46 mV (control) and -23.00±0.59 mV with verapamil (P<0.05, 6 oocytes), with no significant change in slope factors (8.6±0.28 mV and 8.1±0.53 mV for the control and verapamil, respectively, P>0.05). For the Y652A HERG channel, the half-point activation value was -20.29±0.21 mV (control) and -22.10±0.43 mV with verapamil (P<0.05, 6 oocytes), again with no significant change in slope factors (7.98±0.12 mV and 7.54±0.32 mV for the control and verapamil, respectively, P>0.05).
The voltage dependence of WT and the Y652A HERG tail current block by verapamil are shown respectively in Figure 2I, 2J, where the relative tail current represents the ratio of the peak tail currents measured in the presence and absence of verapamil. The steady-state reduction of the WT HERG current by 10 µmol/L verapamil varied as a function of test potential, with the fractional decrease varying from 0.5 at -50 mV to 0.62 at +20 mV (Figure 2I). To achieve equivalent blockade, we used 80 µmol/L verapamil for Y652A. The steady-state block of Y652A HERG was also voltage dependent and varied from 0.48 at -50 mV to 0.63 at +20 mV (Figure 2J). Verapamil blocked Y652A HERG channels only after opening the activation gate. That is consistent with WT HERG. For both channels, the blockade of the HERG current by verapamil increased significantly over the voltage range where HERG channels activated and saturated at voltages where HERG activation was maximal. These results are consistent with the open channel blockade.
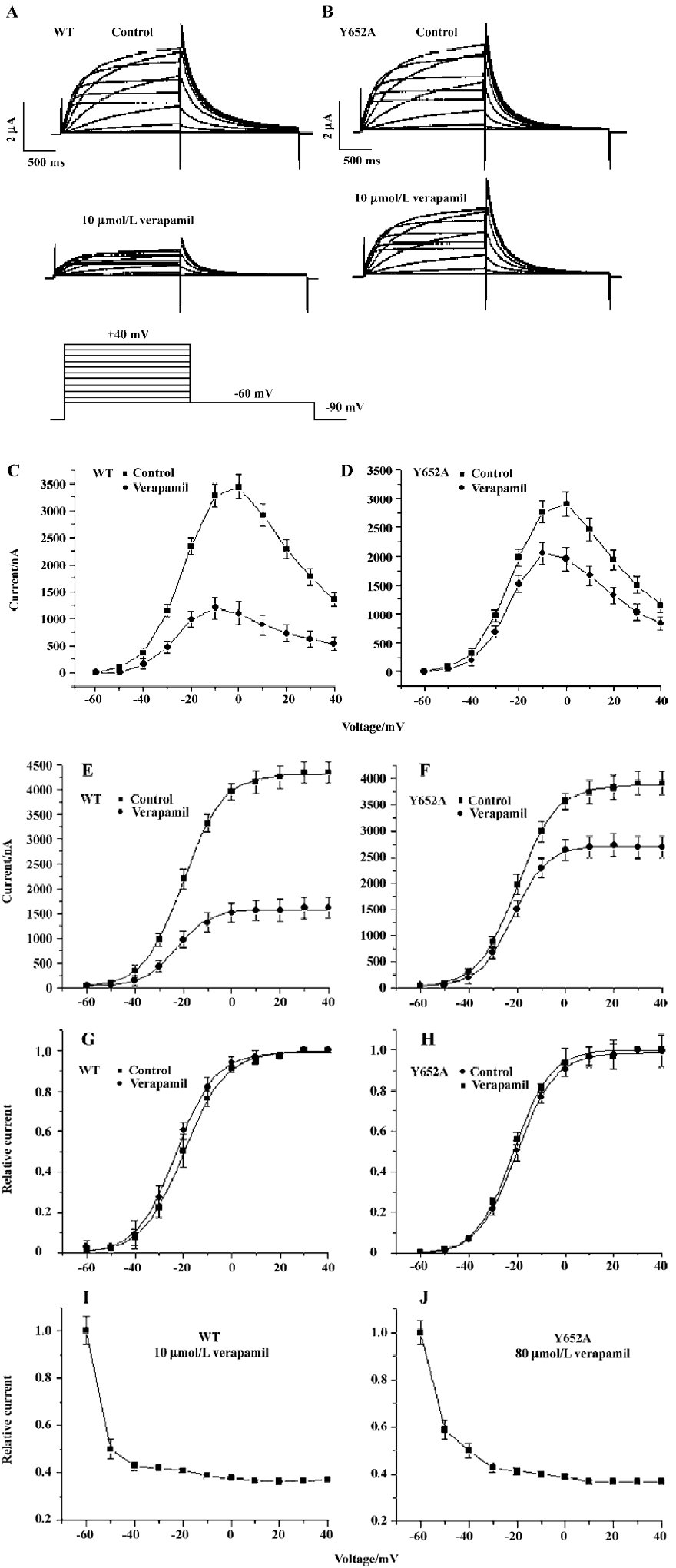
To investigate the state-dependence of the HERG channel blockade by verapamil, a single long test pulse from -90 mV to 0 mV (4000 ms) was used. After the control measure-ment, HERG channels were kept in the closed state at a potential of -90 mV, and 10 µmol/L verapamil was perfused into the bath for 10 min to allow for the equilibration of drug concentrations within the bath and the cell. Then the step protocol was repeated and recorded (n=6, Figure 3A). By the division of currents and presentation in a normalized form, the time course of relative block was obtained (Figure 3B). The fractional block increased rapidly following membrane depolarization, but reached a plateau after 1 s, indicating time dependence of inhibition upon depolarization. This observation argues against closed-channel inhibition and for a dependence of IHERG blockade on channel gating.
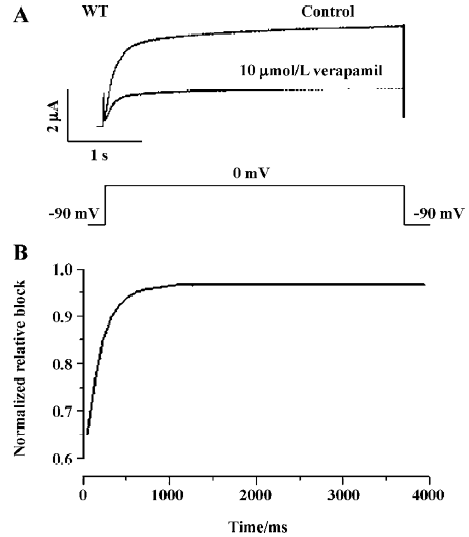
Inactivation of WT and Y652A HERG channels by verapamil The effect of verapamil on the voltage dependence of the inactivation of WT and the Y652 HERG current was assessed using the 3-step protocol[13] shown in the inset of Figure 4C. The steady-state inactivation curve was fitted to a Boltzmann equation (Figure 4C, 4D). Peak current amplitudes were prominently reduced by verapamil (10 µmol/L). For WT HERG channels, the half-point inactivation value was -27.17±4.58 mV (control) and -48.31±6.10 mV with verapamil (P<0.001, 6 oocytes), with no significant change in slope factors (55.9±10.24 and 51.68±8.38 mV for the control and verapamil, respectively, P>0.05, n=6). For the Y652A HERG channels, compared with the negative shift of steady state activation curves by -2 mV, the steady-state inactivation curves also were shifted to more negative values by -14 mV. The half-point inactivation value was -30.38±3.69 mV (control) and -44.19±5.68 mV with verapamil (P<0.001, 6 oocytes), again with no significant change in slope factors (57.66±8.74 and 59.0232±10.187 mV for the control and verapamil, respectively, P>0.05; n=6).
Concentration-dependence blockade of WT and mutant HERG channels by verapamil Previous reports have shown that the mutation of Y652 and F656 to A in the pore helix reduced the blockade of HERG by MK-499, a methane-sulfonanilide antiarrhythmic drug[8]. Mutation of the S6 residues also greatly reduced channel blockade by other compounds such as terfenadine, cisapride, and chloroquine[8,14]. Therefore, we determined the concentration-effect relationship for verapamil on Y652A and F656A HERG channels and compared the potency for blockade with that of the WT HERG channel. Representative traces were recorded in an experiment in which a range of concentrations of verapamil were applied sequentially in the same cell. Current amplitudes were monitored during the control periods and 10 min of each drug application with the same voltage protocol. A total of 5 different concentrations of verapamil were tested. The protocol used to study the effects of verapamil on WT and Y652A IHERG is shown in Figure 5B (lower trace). From a holding potential of -90 mV, the membrane potential was stepped to 0 mV for 2 s, and then repolarized to -60 mV for 2 s before returning to -90 mV. The effect of verapamil on the WT and Y652A HERG channel current is shown in Figure 5A, 5B. Data from individual cells were pooled to obtain the mean (±SEM) fractional block values, which were then plotted against the corresponding verapamil concentration as shown in Figure 5C. Values calculated for tail currents were then fitted with a Hill equation. The half-maximal IC50 was 5.1±1.2 µmol/L for WT and 79.6±16 µmol/L for Y652A.
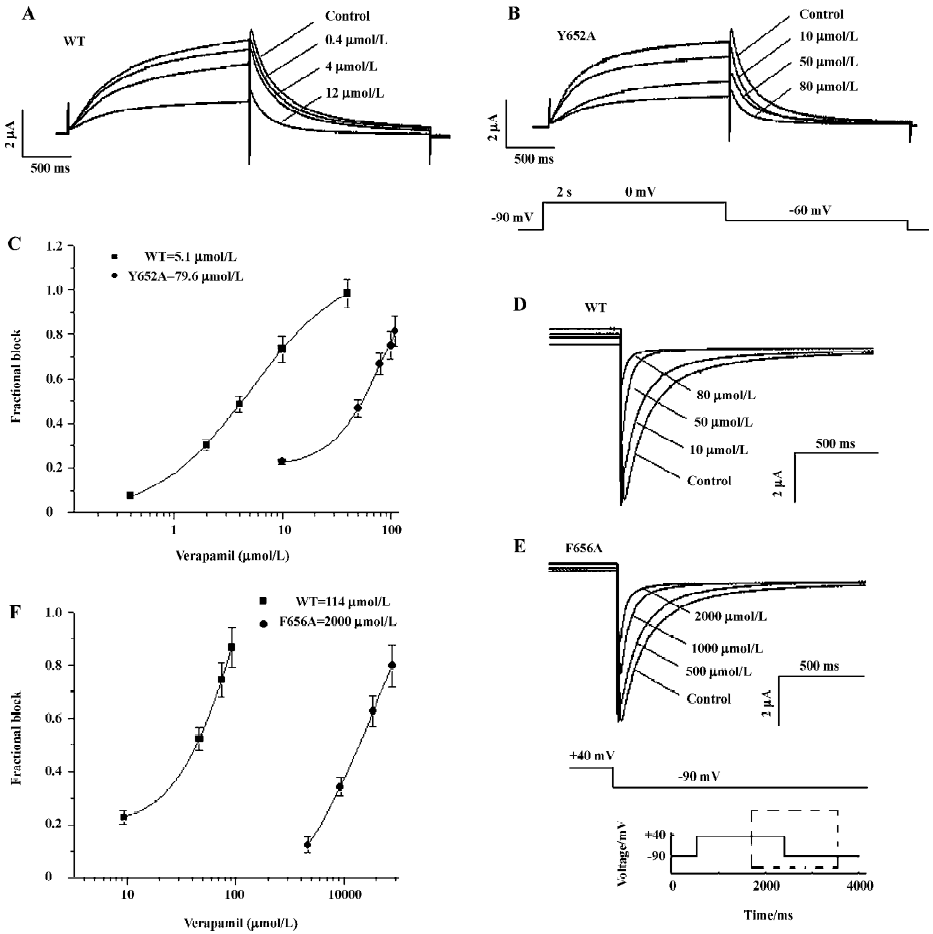
To increase the amplitude of poorly expressed F656A mutant channels for both WT and F656A, 30 mmol/L extracellular K+ were used, and tail currents were recorded at -90 mV (Figure 5D, 5E). Using this protocol, the IC50 was 114±29 µmol/L for the WT current and 2000±120 µmol/L for the F656A HERG current (Figure 5F). Thus, mutation of F656A caused a 20-fold reduction in drug potency, whereas mutation of Y652A reduced potency by a factor of approximately 16. Verapamil blockade was relieved slowly and incompletely upon removal of verapamil. Control values were not reached even after wash-out times of more than 10 min (n=6 oocytes).
Discussion
The cardinal feature of this study is that verapamil preferentially binds to and blocks open HERG channels, and we indicate for the first time that 2 aromatic residues (Tyr-652 or Phe-656) located in the S6 domain of HERG are critical in the verapamil-binding site.
Blockade of HERG channels by verapamil In defining the biophysical properties of the verapamil blockade of WT and the Y652A HERG channel expressed in Xenopus oocytes, we found that the midpoint potential (V0.5) of the activation curves and the inactivation curves were shifted in the hyperpolarizing direction, indicating that verapamil altered both channels; the voltage-dependence of both activation and inactivation gating of the HERG channel shifted to more negative values. In addition, the degree of channel blockade directly correlates with channel activation and verapamil seems to be an open channel blocker of the HERG channel[9]. In our study, the fractional block increased rapidly following membrane depolarization, but reached a plateau after 1 s, indicating time-dependence of inhibition upon depolarization (Figure 3). This is also evident in Figure 2 where the blockade increased significantly over the voltage range (negative to -20 mV) and the HERG channel activated and became saturated at voltages positive to +20 mV (eliciting maximal HERG channel activation). These results suggest that channel activation is required for verapamil blockade, and channel inactivation has little effect on drug affinity.
Previous reports have shown that the voltage dependence for the blockade of HERG channels by chloroquine was reversed by the Y652A mutation[14]. In their study, the blockade of the WT HERG current by chloroquine was enhanced by progressive depolarization. In contrast, the blockade of the Y652A HERG current by this drug was diminished by increased depolarization. These findings suggest that the interaction of chloroquine with the phenol of Y652 mediates a voltage-dependent blockade of WT HERG channels. However, in our study, we found that Y652A did not alter the voltage-dependent block of HERG channels. The blockade of the WT HERG current by verapamil was enhanced by progressive depolarization. Similarly, the blockade of the Y652A HERG current by verapamil was enhanced by increas-ed depolarization (Figure 2I, 2J).
To examine whether key molecular determinants of HERG blockade for previously investigated drugs are also important for HERG channel block by verapamil, we compared the potency of channel block for WT and 2 mutant HERG channels (Y652A and F656A) expressed in oocytes. Verapamil blocked the WT HERG current in a concentration-dependent manner with an IC50 value of 5.1 µmol/L at 0 mV (Figure 5). The potency of channel blockade by verapamil was dramatically reduced in 2 HERG mutant channels (IC50 was increased by 16-and 20-fold at 0 mV for Y652A and F656A, respectively). Our results are consistent with previously reported studies in which all tested HERG-blocking drugs (except fluvoxamine) have been shown to block the mutant HERG channel (F656A) with potency being reduced by over 100-fold, compared to the blockade of the WT HERG channel. Similarly, mutation of Tyr-652 to alanine also leads to dramatic attenuation of HERG blockade by most studied drugs except vesnarinone and fluvoxamine[8,14–17]. As already stated, 2 amino acids located on the S6 transmembrane domain (Y652 and F656) are demonstrated to be important in high-affinity blockade for a number of compounds[8,18]. Other voltage-gated K+ channels (Kv1–Kv4) have Ile and Val (Ile) in the equivalent positions of the aromatic residues Y652 and F656 of HERG. In 2000, Mitcheson and colleagues showed that MK-499 interacted with the HERG channel, and that electrostatic interactions between π electrons and hydrogen atoms of the aromatic rings of Y652/F656 and the drug molecule were crucial for high-affinity binding[12]. Similarly, in our study, for alanine-scanning mutagenesis of Y652 and F656, the potency of channel block by verapamil was dramatically reduced. These results indicate that Tyr-652 and Phe-656 are critical for the verapamil-induced blockade of the HERG channel. These findings suggest a possible structural explanation as to how so many commonly used medications block HERG, but not other Kv channels and should facilitate the rational design of drugs devoid of HERG channel-binding activity.
A previous study reported that the sensitivities of both WT HERG and an inactivation deficient mutant to the methanesulphonanilide E-4031 were lowered by a similar amount of high (K+)e[19]. The authors concluded this effect was likely to be due to a direct interaction: electrostatic repulsion or a “knock-off” process[19]. It seems reasonable to propose that a similar mechanism accounts for the observations regarding the effect of 30 mmol/L (K+)e in Figure 5D of the present study.
Clinical significance It is widely believed that most drugs associated with Torsade de pointes in humans are also associated with the HERG K+ channel blockade at concentrations close to or superimposed upon the free plasma concentrations found in clinical use[20]. Verapamil, an L-type calcium antagonist, blocks native and cloned L-type Ca2+ channels with IC50 values ranging from 250 nmol/L to at least 15.5 µmol/L[21]. It is also reported that verapamil blocks the WT HERG channels in a concentration range similar to that required for the blockade of L-type Ca2+ channels[10]. WT HERG is expressed in HEK 293 cells with an estimated IC50 of 143.0 nmol/L[10], and 3.8±0.2 µmol/L expressed in oocytes[11]. Here in our study, verapamil blocked the WT HERG current in a concentration-dependent manner with an IC50 value of 5.1 µmol/L at 0 mV (Figure 5). The reduced potency of the drug blockade in oocytes compared with mammalian cells is a common finding and may be related to the lipophilic yolk sac in oocytes that acts to sequester the drug[4,16]. However, vera-pamil does not cause QT prolongation in clinical studies. This could be explained by its multiple interactions with cardiac ion channels. That is, verapamil, which does not cause long-QT syndrome, may counteract the potential of the HERG channel blockade which induces QT prolongation early after depolarization generation through its blockade of L-type Ca2+ channels.
To the best of our knowledge, verapamil has been investigated for the first time in our studies that have been designed to examine the importance of the molecular interactions between the key S6 residues and drug molecules. A large number of drugs have been shown to block the HERG channel current[4,5]. So far, however, the HERG channel-binding site has only been investigated for a small number of drugs[8,14–17,22]. More data (well-characterized HERG blockers) are needed to implement the database, which can be used to generate a pharmacophore model[23]. The pharmacophore model may be useful in the pre-synthetic virtual screening of discovery compounds for HERG activity. Although our data provides some insights into the mechanisms of interactions between various drugs and HERG channels, there are still some limitations in our study. To answer the question as to why drugs with diverse structure could inhibit IHERG, extensive site-directed mutagenesis is required in further studies.
Acknowledgements
We are grateful to Dr Michael SANGUINETTI (Utah University) for the HERG clone.
References
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA 1994;91:3438-42.
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995;81:299-307.
- Roden DM. Current status of class III antiarrhythmic drug therapy. Am J Cardiol 1993;72:44B-9.
- Cavero I, Mestre M, Guillon JM, Crumb W. Drugs that prolong QT interval as an unwanted effect: assessing their likelihood of inducing hazardous cardiac dysrhythmias. Exp Opin Pharma-cother 2000;1:947-73.
- De Ponti F, Poluzzi E, Cavalli A, Recanatini M, Montanaro N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes. Drug Safety 2002;25:263-86.
- January CT, Gong Q, Zhou Z. Long QT syndrome: cellular basis and arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol 2000;11:1413-8.
- Tseng GN. I (Kr): the hERG channel. J Mol Cell Cardiol 2001;33:835-49.
- Mitcheson JS, Perry MD. Molecular determinants of high-affinity drug binding to hERG channels. Curr Opin Drug Discov Dev 2003;6:667-74.
- Chouabe C, Drici MD, Romey G, Barhanin J, Lazdunski M. HERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol Pharm 1998;54:695-703.
- Zhang S, Zhou ZF, Gong QM, Makielski JC, Craig T. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res 1999;84:989-98.
- Waldegger G, Niemeyer K, Mörike CA, Wagner H, Suessbrich AE, Busch F, et al. Effect of verapamil enantiomers and metabolites on cardiac K+ channels expressed in Xenopus oocytes. Cell Physiol Biochem 1999;9:81-9.
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 2000;97:12329-33.
- Su Z, Martin RL, Cox BF, Gintant GA. Ziprasidone: an open channel blocker of human ether-a-go-go-related gene K+ channel. J Mol Cell Cardiol 2004;36:151-60.
- Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem 2002;277:23587-95.
- Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high-affinity dofetilide binding to HERG expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol 2000;57:367-74.
- Amiya K, Mitcheson JS, Yasui K, Kodama I, Sanguinetti MC. Open channel block of HERG K+ channels by vesnarinone. Mol Pharmacol 2001;60:244-53.
- Sanchez-Chapula JA, Ferrer T, Navarro-Polanco RA, Sanguinetti MC. Voltage-dependent profile of human ethera-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol Pharmacol 2003;63:1051-8.
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation FEBS Lett 1997;417:43-7.
- Sanguinetti MC, Mitcheson JS. Predicting drug-HERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci 2005;26:119-24.
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 2003;58:32-45.
- Hosey MM, Lazdunski M. Calcium channels: molecular pharma-cology, structure and regulation. J Membr Biol 1988;104:81-105.
- Scholz EP, Zitron E, Kiesecker C, Lueck S, Kathofer S, Thomas D, et al. Drug binding to aromatic residues in the HERG channel pore cavity as possible explanation for acquired long QT syndrome by antiparkinsonian drug budipine. Naunyn Schmiedebergs Arch Pharmacol 2003;368:404-14.
- Cavalli A, Poluzzi E, De Ponti F, Recanatini M. Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K+ channel blockers. J Med Chem 2002;45:3844-53.