
Synaptic plasticity, AMPA-R trafficking, and Ras-MAPK signaling1
Introduction
Long-term synaptic plasticity, the sustained synaptic modification after brief periods of repetitive synaptic activity, was first discovered in the hippocampus[1]. This phenomenon has since been the subject of intense investigation because it was immediately recognized as an experimental model for understanding human cognitive behavior, learning, and memory[2,3]. Although many types of synaptic plasticity have been described, N-methyl-D-aspartate (NMDA)-sensitive glutamate (Glu) receptor (R)-dependent forms of synaptic plasticity in the hippocampal CA1 region remain the most extensively studied. Over the past three decades, significant progress has been made in understanding the cellular and molecular mechanisms underlying these forms of synaptic plasticity[4–9]. Here, we attempt to highlight the progress with emphasis on the last ten years.
Cellular and synaptic mechanisms
The initial studies on synaptic plasticity focused on its cellular mechanisms[2]. These studies revealed that the activation of NMDA-Rs and an influx of calcium ions through the receptor channels are two key processes that trigger synaptic plasticity[2,3]. The activation of NMDA-Rs requires both depolarization and glutamate binding, which explains the two basic properties of synaptic plasticity: input-specificity and input-associativity. Input-specificity means that only synapses activated by repetitive activation can be modified, whereas other synapses on the same cell are normally not modified. This is due to the requirement of glutamate binding for the activation of NMDA-Rs. Input-associativity means that neighboring even weakly-activated synapses can be modified if co-activated with other synapses because the summated depolarization meets the threshold for opening NMDA-Rs.
After resolving the basic cellular mechanisms for synaptic plasticity, much work has been directed to understanding whether the modification occurs on the pre- or postsynaptic sites of synapses, generating a vigorous, highly visible debate for over a decade[3]. The most convincing evidence lands on the postsynaptic side of synapses[10]. However, the discovery of silent synapses and the activation of silent synapses by long term potentiation (LTP) by Malinow and colleagues[11], and later confirmed by many other laboratories[12–17], largely ends the debate. These studies indicate that synaptic plasticity can change the amount of silent synapses, suggesting a simple postsynaptic model that unifies many of the previously conflicting observations.
Synaptic AMPA-R trafficking
The silent synapse theory suggests synaptic trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-sensitive GluRs as a promising mechanism for synaptic modification of transmission efficacy. AMPA-Rs are tetrameric proteins[18,19], which are composed of GluR1, GluR2, GluR2L, GluR3, GluR4, or GluR4c, 6 subunits that are coded by 4 genes, GluR1–4[20–22]. The imaging of green fluorescent protein (GFP)-tagged recombinant GluR1 receptors with 2-photon microscopy has provided the first evidence that AMPA-Rs move into dendritic spines, synaptic sites, during synaptic potentiation[23]. Subsequent studies have revealed that the cytoplasmic carboxyl termini of the constituent subunits, which can be either long or short, govern the synaptic trafficking characteristics of AMPA-Rs[7,24]. So far, multiple distinct synaptic AMPA-R trafficking events have been characterized (Figure 1).
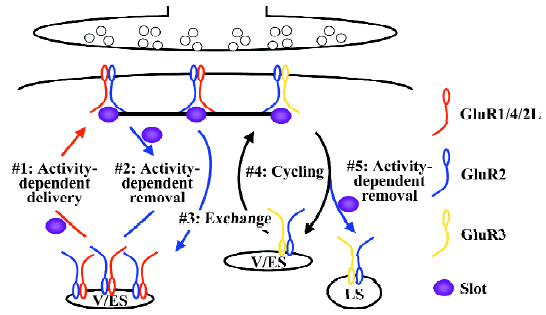
AMPA-Rs with long cytoplasmic termini (ie GluR1-, GluR2L- or GluR4-containing AMPA-Rs) are normally restricted from synapses (Figure 1). During synaptic potentiation (eg long-term potentiation or LTP), synaptic activity activates NMDA-Rs and drives GluR1-, GluR2L-, or GluR4-containing AMPA-Rs into synapses[25–27]. The synaptic delivery of GluR1- and GluR2L-containing AMPA-Rs can be a rapid process, taking approximately 15–25 min during LTP[27,28]. During LTP, GluR1-containing AMPA-Rs are transported to the plasma membrane via exocytosis from recycling endosomes[29]. In hippocampal CA1 neurons, GluR4 is expressed only in the first postnatal week[25]. Spontaneous activity delivers GluR4-containing AMPA-Rs into synapses, which mediates synaptic potentiation during early development[25,30]. The expression of GluR2L peaks at the end of the second postnatal week and declines by half in adults[27]. The synaptic delivery of GluR2L-containing AMPA-Rs requires spontaneous activity[27]. GluR1 expression increases with increasing age and reaches a maximal expression level after the third postnatal week[25]. Strong synaptic activity, such as LTP-inducing stimuli, is required to drive GluR1-containing AMPA-Rs into synapses[26,31]. In the intact brain, experience-independent, spontaneous activity is sufficient to drive GluR2L- and GluR4-containing AMPA-Rs into synapses, whereas experience-dependent activity and/or the presence of other neuromodulatory physiological factors (eg neuromodulators, hormones, and neurotrophic factors) are required for the synaptic delivery of GluR1-containing AMPA-Rs[32–34]. An intriguing hypothesis for how transmission efficacy is maintained despite continuous AMPA-R trafficking and protein turnover involves the simultaneous delivery of slot proteins or slot protein complexes containing AMPA-Rs with long cytoplasmic termini during synaptic potentiation[35]. A recent study has provided evidence supporting the existence of slot proteins and the idea that the hypothesized slot proteins code synaptic transmission strength[36].
Within a short window (approximately 0.5–2 h) after synaptic potentiation, some of the newly-delivered AMPA-Rs with long cytoplasmic termini can be removed from synapses (Figure 1). This process requires synaptic activity and the activation of NMDA-Rs, and mediates depotentiation[37]. The synaptic removal of GluR2L- and GluR1-containing AMPA-Rs occurs rapidly and the process takes approximately 15–25 min[37]. Most likely, the slot proteins are removed together with AMPA-Rs with long cytoplasmic termini in order to reduce transmission efficacy[36].
Other synaptic AMPA-Rs with long cytoplasmic termini (ie those not removed by depotentiation) are eventually exchanged with AMPA-Rs with only short cytoplasmic termini (Figure 1). The synaptic exchange of GluR1-, GluR2L-, and GluR4-containing AMPA-Rs with GluR2-containing AMPA-Rs (ie GluR2/3 AMPA-Rs) requires no synaptic activity[25,27,36]. This process has a slow rate time constant of approximately 16 h and is essential for maintaining the capacity for bidirectional plasticity[36]. The slot proteins play a key role in maintaining transmission strength during this slow exchange[36].
AMPA-Rs with only short cytoplasmic termini, constitutively cycle between synaptic and non-synaptic sites (Figure 1). The synaptic cycling of GluR2-containing AMPA-Rs (ie GluR2/3 AMPA-Rs) has a rapid time rate(time constant of approximately 15–20 min) and the process requires no synaptic activity[38–42]. Recent studies suggest that most of the cycling GluR2-containing AMPA-Rs may bind to slot proteins at the synaptic site, and the pool of cycling GluR2-containing AMPA-Rs at the non-synaptic site is most likely very small[36,43]. Together, synaptic AMPA-R exchange and cycling serve to maintain synaptic strength despite continuous protein turnover.
AMPA-Rs with only short cytoplasmic termini can be removed from synapses (Figure 1). During synaptic depression [eg long-term depression (LTD)], synaptic activity activates NMDA-Rs and removes GluR2-containing AMPA-Rs (ie GluR2/3 AMPA-Rs)[28,44–46]. The synaptic removal of GluR2-containing AMPA-Rs can be a rapid process, taking approximately 15–25 min during LTD[28], and the receptors are diverted to the late endosomes/lysosomes via clathrin-dependent endocytosis[47–50]. The slot proteins are most likely removed together with GluR2-containing AMPA-Rs, which accounts for the reduced transmission efficacy after LTD[36].
GluR2-lacking AMPA-Rs may also traffic into synapses in physiological and pathological conditions[51]. Although the endoplasmic reticulum (ER) normally only allows properly assembled AMPA-Rs to exit, and the majority of AMPA-Rs that exit from the ER contain GluR2 subunits[19], some endogenous GluR2-lacking AMPA-Rs do egress from the ER and travel into synapses[25,52,53]. For example, during early postnatal development, the expression of GluR2 is relatively low[25,27], and GluR2-lacking AMPA-Rs may mediate a significant portion of synaptic transmission[25,54]. In juvenile and adult neurons, the synaptic presence of GluR2-lacking AMPA-Rs is regulated by the synaptic trafficking of GluR2-containing AMPA-Rs, which are controlled by an interaction with protein kinase C (PKC)-interacting protein 1 (PICK1) or glutamate receptor-interacting protein (GRIP1, also called AMPA-binding protein)[55,56]. In the normal condition, the number of GluR2-lacking AMPA-Rs at synapses is limited due to synaptic exchange and replacement by GluR2-containing AMPA-Rs[36,53,57,58]. Because GluR2-lacking AMPA-Rs are permeable to calcium, their synaptic presence is often associated with pathological conditions, such as brain ischemia and amyotrophic lateral sclerosis[59–61].
Synaptic Ras/MAPK signaling
The heavy, regulated trafficking of AMPA-Rs at synapses implicates the existence of a trafficking control system. Ras family small GTPase–mitogen-activated protein kinase (MAPK) signaling pathways are ideal candidates for signaling synaptic AMPA-R trafficking events for several reasons. First, these signaling pathways are known to control a variety of intracellular processes[62–64]. In addition, Ras family small GTPases Ras, Rap1, and Rap2, as well as their upstream regulators and downstream effectors, including p42/44 MAPK, phosphoinositide 3 kinase (PI3K), c-Jun amino-terminal kinase (JNK), and p38 MAPK, are expressed at synapses[6,9,65]. Finally, diseases causing cognitive impairment are associated with genetic defects of molecules involved in Ras-MAPK signaling (eg calcineurin with schizophrenia[66], H-Ras with autism[67], p38 MAPK, and JNK with Alzheimer’s disease[68,69], B-Raf with cardio-facio-cutaneous (CFC) syndrome[70], RasGap neurofibromin with neurofibromatosis type 1 (NF1)[71,72], tuberin with tuberous sclerosis[73], and Rsk with Coffin-Lowry syndrome and X-linked mental retardation[74,75]. In support of this notion, recent findings have shown that different Ras-MAPK signaling pathways differentially control synaptic trafficking of AMPA-Rs during distinct forms of synaptic plasticity.
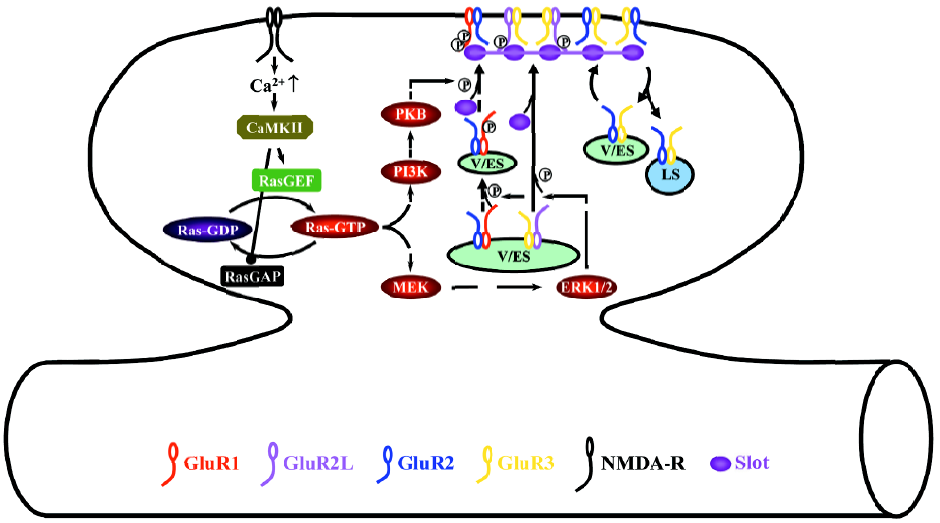
During LTP, the activation of NMDA-Rs stimulates small GTPase Ras-extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK, also named p42/44 MAPK), PI3K, and protein kinase B (PKB, also called Akt) signaling pathways (Figure 2)[28,34,76]. Different signaling molecules relay the activation of NMDA-Rs to Ras at different developmental stages, whereas neonatal neurons require cyclic AMP-dependent protein kinase A (PKA) and Ras activator son of sevenless and juvenile and adult neurons need calcium/calmodulin-dependent protein kinase II and Ras activator Ras-guanyl-nucleotide releasing factor (GRF)[25,77,78]. The activation of the Ras-MEK-ERK pathway stimulates phosphorylation of S841 of GluR2L and S845 of GluR1, whereas the activation of the Ras-PI3K-PKB pathway stimulates phosphorylation of S831 of GluR1[34]. S841 phosphorylation of GluR2L is sufficient to drive GluR2L-containing AMPA-Rs into synapses, while phosphorylation of both S845 and S831 of GluR1 is required for the synaptic delivery of GluR1-containing AMPA-Rs[34]. Because ERK and PKB are unlikely to directly phosphorylate GluR1 and GluR2L, other molecules must exist at synapses to relay the signaling[63,64]. The likely candidates include PKA[79], PKC[80,81], ribosomal S6 kinase[74,75,82], and the mammalian target of rapamycin[83,84]. Determining the precise functional relationships (ie sequential or parallel, and downstream or upstream) of the signaling molecules involved in Ras pathways during LTP will be central to future studies[6].
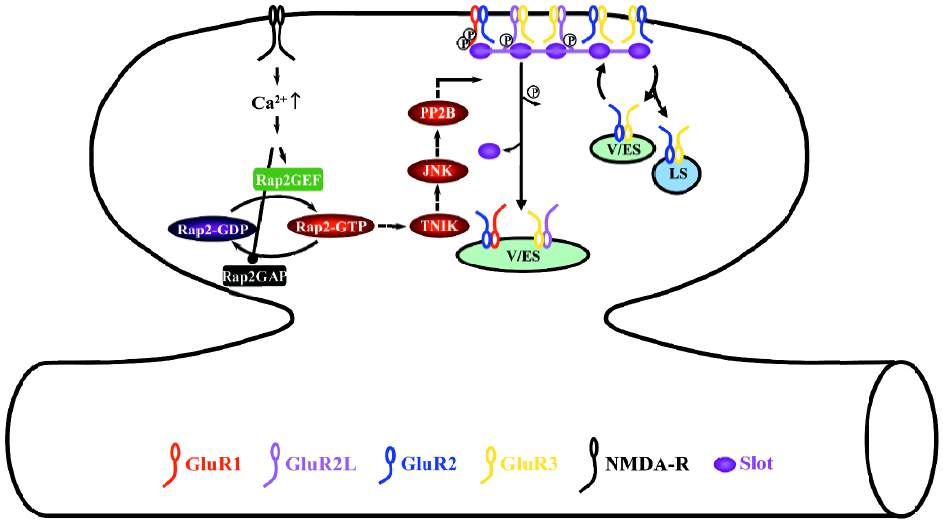
During depotentiation, the activation of NMDA-Rs stimulates the small GTPase Rap2-Traf2 and NCK-interacting kinase (TNIK)-JNK signaling pathway (Figure 3)[37]. The activation of the Rap2-TNIK-JNK pathway dephosphorylates S841 of GluR2L, and S845 and S831 of GluR1, which removes GluR2L- and GluR1-containing AMPA-Rs from synapses during depotentiation[37]. Dephosphorylation seems to be mediated by protein phosphatase 2B (also named calcineurin) downstream of Rap2-TNIK-JNK[37], which is consistent with the finding that calcineurin mediates depotentiation[85–87].
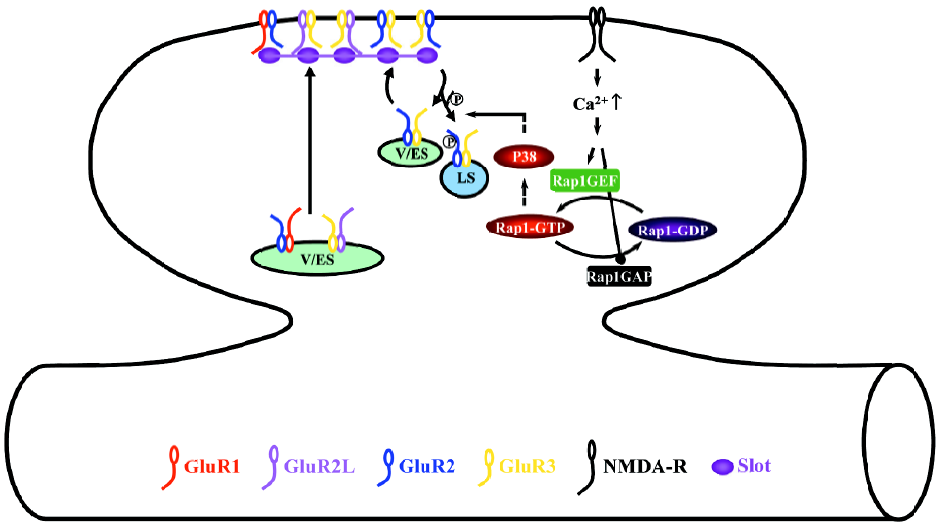
During LTD, the activation of NMDA-R stimulates the small GTPase Rap1-p38 MAPK signaling pathway (Figure 4)[28]. Phosphorylation of S880 of GluR2 modulates the interaction of GluR2 with GRIP1 and PICK1[46,88,89], which controls the synaptic anchoring of GluR2-containing AMPA-Rs[90–96]. Thus, the activation of the Rap1-p38 MAPK pathway most likely controls the synaptic removal of GluR2-containing AMPA-Rs during LTD via regulating phosphorylation of S880 of GluR2[28,44,46]. Because p38 MAPK does not phosphorylate AMPA-Rs directly[63,97], other synaptic signaling molecule(s), such as PKC[88,89,92] and/or MAPK-interacting kinase[98], may relay the signaling and phosphorylate GluR2.
Conclusion
In the last decade, accumulating evidence indicates that postsynaptic trafficking of AMPA-Rs plays a key role in regulating synaptic transmission and plasticity. Multiple Ras-MAPK signaling pathways control the regulated synaptic trafficking of AMPA-Rs during different forms of synaptic plasticity. However, a few fundamental issues remain unsolved, for example, different populations of synapses in individual neurons having distinct AMPA-R compositions. How AMPA-Rs recognize and traffic into different types of synapses remains puzzling. Also, many of the regulators in Ras-MAPK signaling pathways have poor selectivity in interacting with their effectors. How different Ras-MAPK pathways differentially signal trafficking of distinct pools of AMPA-Rs at single synapses is still elusive. More impor-tantly, how synaptic AMPA-R trafficking and Ras-MAPK signaling are regulated in different behavioral states in the intact brain remains largely unknown. One speculation is that AMPA-R trafficking and Ras-MAPK signaling are compartmentalized between and within synapses, and compartmentalized trafficking and signaling are differentially regulated in different behavioral states. Resolving the subcellular compartmental protein trafficking and signaling in these physiological conditions demands high-resolution experimental techniques. Several recently-refined techniques, including multiple whole-cell in vivo recordings[99–103], real time single spine Ras/kinase activity monitoring[104,105], living cell imaging and immunogold electron microscopy[106–110], when combined with a recombinant DNA in vivo delivery technique[32–34,36,111], provide powerful high-resolution approaches that may resolve these pivotal issues.
Acknowledgements
We thank the members of the Zhu Laboratory for helpful comments and discussions. In addition, we apologize to those whose work we did not cite because of space limita-tions.
References
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331-56.
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993;361:31-9.
- Malenka RC, Nicoll RA. Long-term potentiation –– a decade of progress? Science 1999;285:1870-4.
- Malinow R. AMPA receptor trafficking and long-term potentia-tion. Philos Trans R Soc Lond B Biol Sci 2003;358:707-14.
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 2004;44:5-21.
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 2004;5:173-83.
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 2004;5:952-62.
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science 2006;311:1253-6.
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol 2006;16:95-101.
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol 1993;70:1451-9.
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 1995;375:400-4.
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron 1995;15:427-34.
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature 1996;381:71-5.
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron 1997;18:269-80.
- Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, et al. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci 1999;2:972-7.
- Liao D, Zhang X, O’Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci 1999;2:37-43.
- Petralia RS, Esteban JA, Wang YX, Partridge JG, Zhao HM, Wenthold RJ, et al. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat Neurosci 1999;2:31-6.
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science 1998;280:1596-9.
- Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron 2003;40:763-74.
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci 1994;17:31-108.
- Schoepfer R, Monyer H, Sommer B, Wisden W, Sprengel R, Kuner T, et al. Molecular biology of glutamate receptors. Prog Neurobiol 1994;42:353-7.
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 1999;51:7-61.
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 1999;284:1811-6.
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 2002;25:103-26.
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 2000;3:1098-106.
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 2000;287:2262-7.
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, et al. Glutamatergic plasticity by synaptic delivery of GluR-B (long)-containing AMPA receptors. Neuron 2003;40:1199-212.
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 2002;110:443-55.
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science 2004;305:1972-5.
- Zhu JJ, Malinow R. Acute versus chronic NMDA receptor blockade and synaptic AMPA receptor delivery. Nat Neurosci 2002;5:513-4.
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 2001;105:331-43.
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science 2003;299:1585-8.
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science 2005;308:83-8.
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, et al. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev 2005;19:2000-15.
- Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol 2000;10:352-7.
- McCormack SG, Stornetta RL, Zhu JJ. Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron 2006;50:75-88.
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, et al. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron 2005;46:905-16.
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, et al. NSF binding to GluR2 regulates synaptic transmission. Neuron 1998;21:87-97.
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, et al. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP- dependent interaction with NSF and alpha- and beta-SNAPs. Neuron 1998;21:99-110.
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 1998;21:393-400.
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 1999;24:649-58.
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 2002;36:661-74.
- Adesnik H, Nicoll RA, England PM. Photoinactivation of native AMPA receptors reveals their real-time trafficking. Neuron 2005;48:977-85.
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 2003;300:1751-5.
- Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron 2003;39:163-76.
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci 2003;23:9220-8.
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, et al. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 2000;3:1291-300.
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000;25:635-47.
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 2004;43:221-36.
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron 2005;45:81-94.
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca(2+)-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 2006;16:288-97.
- Clem RL, Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 2006;49:663-70.
- Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depres-sion. Nat Neurosci 2006;9:636-41.
- Kumar SS, Huguenard JR. Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J Neurosci 2003; 23: 10 074–83.
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, et al. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron 2005;45:903-15.
- Liu SJ, Cull-Candy SG. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat Neurosci 2005;8:768-75.
- Bagal AA, Kao JP, Tang CM, Thompson SM. Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci USA 2005; 102: 14 434–9.
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term poten-tiation. Nat Neurosci 2006;9:602-4.
- Liu S, Lau L, Wei J, Zhu D, Zou S, Sun HS, et al. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 2004;43:43-55.
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, et al. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J Neurosci 2006;26:11798-806.
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci 2006;26:5309-19.
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev 2001;81:153-208.
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807-69.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489-501.
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 2000;3:661-9.
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA 2003;100:8987-92.
- Comings DE, Wu S, Chiu C, Muhleman D, Sverd J. Studies of the c-Harvey-Ras gene in psychiatric disorders. Psychiatry Res 1996;63:25-32.
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 2002;22:3376-85.
- Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, . Neurotoxic mechanisms caused by the Alzheimer’s disease-linked swedish APP mutation: oxidative stress, caspases and JNK pathway. J Biol Chem 2003; 278: 28 294–302.
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 2006;38:294-6.
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002;415:526-30.
- Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci 2002;5:95-6.
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577-90.
- Yntema HG, van den Helm B, Kissing J, van Duijnhoven G, Poppelaars F, Chelly J, et al. A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics 1999;62:332-43.
- Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, et al. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature 1996;384:567-70.
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 1997; 272: 19 103–6.
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 2003;6:15-6.
- Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA. Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J 2004;23:1567-75.
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 2003;6:136-43.
- Blackstone C, Murphy TH, Moss SJ, Baraban JM, Huganir RL. Cyclic AMP and synaptic activity-dependent phosphorylation of AMPA- preferring glutamate receptors. J Neurosci 1994;14:7585-93.
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole- 4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem 1997; 272: 32 528–33.
- Thomas GM, Rumbaugh GR, Harrar DB, Huganir RL. Ribosomal S6 kinase 2 interacts with and phosphorylates PDZ domain-containing proteins and regulates AMPA receptor trans-mission. Proc Natl Acad Sci USA 2005; 102: 15 006–11.
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003;13:1259-68.
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 2002;99:467-72.
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science 1993;261:1051-5.
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, et al. A selective role of calcineurin aalpha in synaptic depotentiation in hippocampus. Proc Natl Acad Sci USA 1999;96:4650-5.
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, et al. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 2001;104:675-86.
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci 2000;20:7258-67.
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron 2000;28:499-510.
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, et al. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron 1998;21:581-91.
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron 1999;22:179-87.
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, et al. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 2000;28:873-86.
- Osten P, Khatri L, Perez JL, Köhr G, Giese G, Daly C, et al. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron 2000;27:313-25.
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA 2001; 98: 11 725–30.
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, et al. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron 2003;37:625-38.
- Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, et al. Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. J Neurosci 2006;26:2380-90.
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol 2002;3:663-72.
- Asaki C, Usuda N, Nakazawa A, Kametani K, Suzuki T. Localization of translational components at the ultramicroscopic level at postsynaptic sites of the rat brain. Brain Res 2003;972:168-76.
- Zhu JJ, Connors BW. Intrinsic firing patterns and whisker-evoked synaptic responses of neurons in the rat barrel cortex. J Neurophysiol 1999;81:1171-83.
- Larkum ME, Zhu JJ. Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci 2002;22:6991-7005.
- Margrie TW, Brecht M, Sakmann B. In vivo, low-resistance, whole-cell recordings from neurons in the anaesthetized and awake mammalian brain. Pflugers Arch 2002;444:491-8.
- Zhu Y, Stornetta RL, Zhu JJ. Chandelier cells control excessive cortical excitation: characteristics of whisker-evoked synaptic responses of layer 2/3 nonpyramidal and pyramidal neurons. J Neurosci 2004;24:5101-8.
- Lee AK, Manns ID, Sakmann B, Brecht M. Whole-cell recordings in freely moving rats. Neuron 2006;51:399-407.
- Helmchen F, Fee MS, Tank DW, Denk W. A miniature head-mounted two-photon microscope. High-resolution brain imaging in freely moving animals. Neuron 2001;31:903-12.
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, Svoboda K. Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci 2006;9:283-91.
- Rubio ME, Wenthold RJ. Glutamate receptors are selectively targeted to postsynaptic sites in neurons. Neuron 1997;18:939-50.
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004;429:761-6.
- Horton AC, Racz B, Monson EE, Lin AL, Weinberg RJ, Ehlers MD. Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 2005;48:757-71.
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 2006;26:2000-9.
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, et al. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 2006;52:817-30.
- Komai S, Licznerski P, Cetin A, Waters J, Denk W, Brecht M, et al. Postsynaptic excitability is necessary for strengthening of cortical sensory responses during experience-dependent development. Nat Neurosci 2006;9:1125-33.