
Cardiac sympathetic nerve terminal function in congestive heart failure1
Introduction
It has been long recognized that cardiac norepinephrine (NE) is depleted in patients with congestive heart failure[1,2]. Early studies have shown that NE depletion is associated with increased release of cardiac NE secondary to heightened sympathetic nervous activity and decreased synthesis of NE in patients with congestive heart failure[2–5]. It was thought initially that the heightened sympathetic activity was an important adaptive mechanism to support the failing myocardium, and that the subsequent depletion of cardiac NE stores contributed to the progressive deterioration of cardiac function and the decreased myocardial contractility seen in chronic heart failure. However, it was later discovered that the intrinsic contractility of the heart muscle remains normal after depletion of myocardial NE by reserpine treatment or cardiac denervation[6]. Thus, normal cardiac stores of NE are not essential for maintaining the intrinsic myocardial contractility, and NE depletion does not account for the myocardial depression in heart failure. However, as an overwhelming majority of NE is stored in the intraneuronal storage vesicles, tissue content of NE does not accurately reflect myocardial interstitial NE concentration which is elevated in heart failure. Recent studies from our laboratory and others have provided new insights into cardiac sympathetic nerve terminal function in heart failure, and it has been suggested that abnormal NE uptake in the sympathetic nerve ending plays an important pathophysiological role in dilated cardiomyopathy. The findings further indicate that the change in NE uptake in chronic heart failure is maladaptive, and might be a novel therapeutic target in the treatment of congestive heart failure.
Reduction of NE uptake transporter in heart failure
Myocardial uptake of NE is known to be reduced in the failing heart. In experimental heart failure produced by aortic constriction, Spann et al[7] showed that the intravenous infusion of NE resulted in a much smaller increase in cardiac NE in guinea pigs with heart failure than normal animals, but the increase of NE in the kidneys did not differ between the 2 groups of animals. They attributed the organ-specific difference of tissue NE uptake to a diminished number of sympathetic nerves and/or binding sites in the failing heart. We now know that the primary defect is caused by a reduction of neuronal NE transporter (NET) density at the sympathetic nerve endings[8]. Since the NE uptake mechanism is responsible for a rapid removal of interstitial NE after the sympathetic release of NE, this defect of NE uptake has been used to explain, at least in part, the selective increase of the cardiac washout of NE. The amount of NE in the myocardial interstitial space is also expected to increase and causes greater actions on the postsynaptic adrenergic receptors. NET, a 617 amino acid protein, comprises of 12 transmembrane domains at the sympathetic nerve endings[9]. It is a member of the Na+ and Cl–-dependent family of neurotransmitter transporters. It takes up NE from the interstitial space back to the adrenergic nerve terminals with the stoichiometric exchange of Na+ and Cl– against their electrochemical gradients[10].
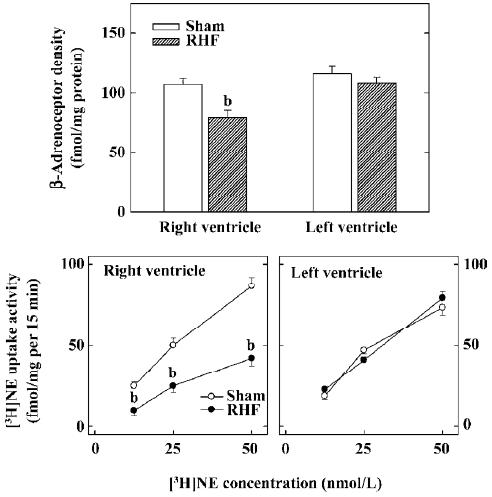
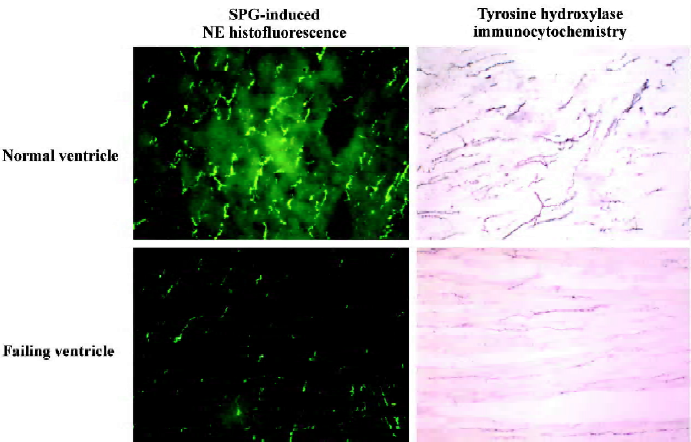
Our laboratory has studied the pre- and postsynaptic function of the cardiac sympathetic nerves for many years. Our interest began with a novel observation in experimental, right ventricle heart failure dogs produced by tricuspid avulsion, and progressive pulmonary artery constriction where myocardial β-receptor density was reduced only in the failing right ventricle[11]. The chamber-specific reduction of myocardial β-receptor density was later confirmed in the failing human right ventricles associated with primary pulmonary hypertension[12]. We speculated that the decrease of myocardial β-receptors occurred because of a chamber-specific reduction of cardiac sympathetic NE uptake activity, leading to an increase of interstitial NE of the failing right ventricular myocardium. Indeed, we have shown that myocardial neuronal NE uptake activity as measured by the tissue accumulation of [3H]NE ex vivo is reduced only in the failing right ventricular myocardium, and this change correlated with the reduction of myocardial β-receptors (Figure 1)[13]. We later studied the cardiac sympathetic nerve terminal dysfunction in heart failure by measuring the NE binding site density and numbers of sympathetic neuronal marker NE and tyrosine hydroxylase, using sucrose-potassium phosphate-glycoxylic acid-induced NE histofluorescence and tyrosine hydroxylase immunocytochemistry, respectively[13,14]. The sympathetic neuronal markers were reduced in the failing ventricle (Figure 2). In contrast, the contralateral non-failing left ventricle is relatively spared without reductions of myocardial β-receptors, NE uptake activity, NE uptake binding sites, or the numbers of neuronal profiles of NE and tyrosine hydroxylase. The NE uptake binding site density also did not change in the kidneys of the heart failure animals. These findings suggest that the changes in cardiac sympathetic nerve endings are produced by a local mechanism, and is organ- and chamber-specific, occurring only in the failing ventricle.
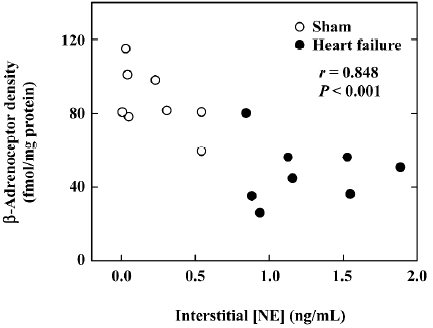
We have extended our observations in right ventricle heart failure animals to dogs with biventricular heart failure produced by rapid ventricular pacing[14]. In these animals, we also produced direct proof that myocardial interstitial NE increased in the failing heart, and that the interstitial NE correlated inversely with number of myocardial β-receptors (Figure 3)[15], indicating that the changes in the myocardial β-adrenoceptor density is agonist induced, secondary to the reduction of NE neuronal uptake.
The functional importance of the NE uptake site was further studied in rabbits at various time intervals after the start of rapid ventricular pacing[16]. We found that rapid ventricular pacing caused early sympathetic nervous system activation, followed in sequence by the reduced myocardial NE uptake, loss of neuronal NE, and downregulation of myocardial β-adrenoceptors. However, there was no significant reduction of the protein gene product 9.5, a panneuronal marker, suggesting that the anatomic integrity of the cardiac sympathetic nerves probably is intact, and the changes of sympathetic neurotransmitters within the nerve endings are caused by functional abnormalities that are potentially reversible with either effective therapy or the removal of a primary insult that causes heart failure. The interdependence of increased sympathetic stimulation, decreased cardiac NE uptake, and myocardial β-adrenoceptor downregulation is further discussed in a study by Leineweber et al[17] who found that neurohumoral activation is essential for the reduction of myocardial β-receptors in the hypertrophied right ventricle produced by monocrotaline. This study, which is similar to our earlier studies of right ventricle heart failure, is characterized by a chamber-specific reduction of myocardial NE uptake sites[18].
Significance of NE re-uptake for cardiac function in heart failure
The physiological significance of the NE re-uptake mechanism in the regulation of myocardial β-receptor density and postsynaptic β-adrenergic inotropic responsiveness was further studied in heart failure animals treated with desipramine[19] and selegiline[20]. Desipramine is a NET inhibitor. In the present study, it increased myocardial interstitial NE in heart failure, and caused further reductions of myocardial β-adrenoceptor density and β-adrenergic subsensitivity. In contrast, selegiline, which is a central α2-agonist with a neuroprotective effect, attenuated the increase in plasma NE and the decrease of myocardial β-receptor density and improved cardiac mechanical function in pacing-induced cardiomyopathy. These findings support the concept that interstitial NE is a modifiable variable, important in the mediation of agonist-induced postsynaptic events seen in heart failure.
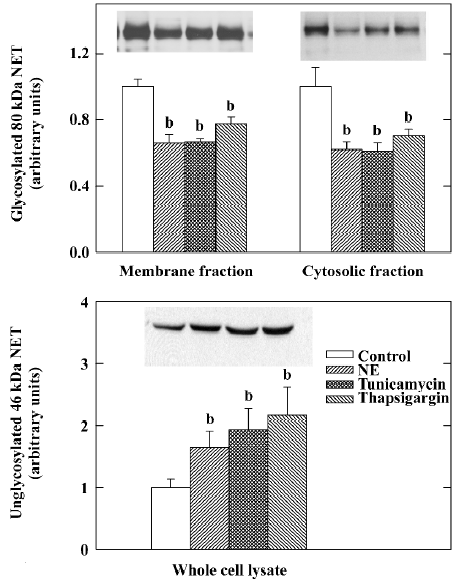
To study the mechanism responsible for the NE uptake inhibition in heart failure, experiments have been conducted in our laboratory to show that the reductions of cardiac sympathetic transmitters and NET can be induced by exogenous NE[21,22] and inhibited by desipramine[19,23] and antioxidants[22,24] in intact animals. Studies also have been conducted in cultured rat neuroblastoma cells (PC12) cells, indicating that NE reduces NE uptake activity and the NET protein in a dose-dependent fashion[25]. The changes of NE on the NET protein are reproduced by well-known endoplasmic reticulum stressors such as tunicamycin and thapsigargin (Figure 4). These effects of NE are most likely caused by endoplasmic reticulum stress, the resultant reduced glyco-sylation and the trafficking of NET to the cell membrane[26]. Our studies[26] have also shown that the reduction of the NET protein by NE in PC12 cells was not associated with changes in NET mRNA, suggesting that the NET protein downregulation is a post-transcriptional event. These findings are consistent with an earlier study of experimental heart failure in which the reduction of the cardiac NET protein in aortic-banded rats was associated with no change of NET mRNA in the left stellate ganglion[27]. NET gene expression also was unchanged in the stellate ganglia of rats with cardiac hypertrophy induced by pressure overload, despite a marked reduction of NE uptake site density in the hypertrophied left ventricle[28]. There is also evidence that the effects of NE on NET are associated with an increase in reactive oxygen species, and can be attenuated by the free-radical scavenger mannitol, or antioxidant enzymes superoxide dismutase and catalase. The findings suggest that the cardiac sympathetic nerve terminal dysfunction is probably caused by increased interstitial NE in heart failure, and the neuronal damage effect of NE involves the uptake of NE or its oxidative metabolites into the sympathetic nerve endings. More recently, endothelin-1 also has been shown to inhibit cardiac NE uptake and promote NE release from the failing heart via ETA receptors[29]. Endothelin blockade also improves survival in experimental heart failure[30,31], but no long-term beneficial effects of either ETA-specific[32] or non-specific endothelin receptor inhibitors[33] have been demonstrated in human heart failure.
Myocardial metaiodobenzylguanidine scintigraphy and its clinical utility in heart failure
Recently, radio-iodinated metaiodobenzylguanidine (MIBG), a structural analogue of NE, has been used to study the integrity and function of the cardiac sympathetic nervous system[34]. MIBG shares the same reuptake mechanism and storage site with NE. Thus, its uptake into the myocardium reflects both the distribution of cardiac sympathetic innervation and the extent of neuronal NE uptake activity. The failing heart is characterized by reduced distribution and washout of MIBG[35]. Abnormal MIBG uptake, calculated by the ratio of heart and mediastinum uptake, correlates with reduced myocardial contractile reserve in patients with dilated cardiomyopathy[36]. Similarly, c-11-HED, a PET-based NE analog, is significantly correlated to the NET density, and has been used to demonstrate regional variations of NE content in cardiomyopathy[37]. Thus, the MIBG and HED-PET patterns can be used as a non-invasive means to investigate the changes of cardiac sympathetic innervation in the hearts of cardiomyopathic patients. Studies have now shown that cardiac sympathetic nerve innerva-tion, as demonstrated by MIBG scintigraphy, is an independent predictor for adverse clinical outcomes, including mortality in patients with heart failure[38,39]. Improvements in MIBG patterns also have been shown to occur in patients who respond favorably to carvedilol[40], metoprolol[41], spironolactone[42,43], enalapril[44], and cardiac resynchronization therapy[45]. In contrast, bucindolol therapy, which showed only marginal survival benefits[46], did not improve the sympathetic nerve function as measured by MIBG[47]. However, because prior studies involved only small numbers of patients, the application of MIBG scintigraphy in heart failure remains investigational. In a recent editorial, Motherwell et al[48] discussed the proper use of MIBG scintigraphy and the limitations of current imaging protocols, as well as need of a better understanding of the kinetics of MIBG and standardization of imaging techniques and analyses in cardiac imaging. They concluded that a large multicenter trial with a standardized imaging protocol is required to establish the clinical utility of the MIBG in congestive heart failure in patients.
Therapeutic implications
Long-term β-blocker therapy is now widely accepted as a pillar in the treatment of systolic heart failure. Effective utilization of β-receptor blockers can not only improve left ventricular systolic function, but also increase survival in patients with chronic heart failure secondary to left ventricular systolic dysfunction[49–52]. Given the overwhelming success of the β-adrenoceptor blocker therapy, attempts have been made to determine if similar or greater beneficial effects can be derived from potent sympatholytic agents such as moxonidine, which has been shown to decrease peripheral sympathetic outflow and circulating plasma NE by stimulating the brain stem imidazoline-1 receptor[53]. Unfortunately, despite early enthusiasm with the centrally-acting sympatholytic agents[54,55], moxonidine therapy was considered detrimental because it tended to increase mortality and morbidity in chronic systolic heart failure in a large clinical trial[56]. Thus, the sympathetic nervous system activation can be both adaptive and maladaptive, depending on the degree of basal sympathetic activation and the extent of sympa-tholysis or β-receptor blockade. Furthermore, generalized sympathetic nervous system inhibition probably has limited therapeutic utility, and localized adrenergic inhibition at the cardiac receptor level is the preferred mode of therapy for heart failure.
Alternatively, the results of several recent studies suggest that without directly affecting the central sympathetic drive, cardiac function in heart failure may be modified by agents or interventions that upregulate the neuronal NET in the myocardium. Kreusser et al[57] reported that an injection of nerve growth factor into the stellate ganglia of rats with heart failure produced by transverse aortic constriction, improved NE uptake, repleted cardiac NE stores, and increased left ventricular fractional shortening. The number of cardiac sympathetic nerves, however, was unaffected. In a separate study[58], adenoviral gene transfer was used to overexpress NET in the myocardium of rabbits with pacing-induced cardiomyopathy. This resulted in increased NE uptake capacity and the reversal of β-receptor downregulation in the cardiac tissue. Local overexpression of cardiac NET also improved the systolic function and contractile reserve of the cardiomyopathic hearts. These findings not only confirm the importance of NET in the initiation or progression of cardiomyopathy, but also suggest that cardiac NET may be a novel therapeutic target in the treatment of congestive heart failure. Future research should be directed at the development of pharmacological agents or interventions that reduce the cardiac noradrenergic drive while preserving the integrity and NE reuptake function of the sympathetic nerve terminals.
References
- Braunwald E. Alterations in the activity of the adrenergic nervous system in heart failure. UCLA Forum Med Sci 1970;10:289-94.
- Chidsey CA, Braunwald E. Sympathetic activity and neurotransmitter depletion in congestive heart failure. Pharmacol Rev 1966;18:685-700.
- Pool PE, Covell JW, Levitt M, Gibb J, Braunwald E. Reduction of cardiac tyrosine hydroxylase activity in experimental congestive heart failure. Its role in the depletion of cardiac norepinephrine stores. Circ Res 1967;20:349-53.
- Dequattro V, Nagatsu T, Mendez A, Verska J. Determinants of cardiac noradrenaline depletion in human congestive failure. Cardiovasc Res 1973;7:344-50.
- Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996;93:1667-76.
- Spann JF, Sonnenblick EH, Cooper T, Chidsey CA, Willman VL, Braunwald E. Cardiac norepinephrine stores and the contractile state of heart muscle. Circ Res 1966;19:317-25.
- Spann JF Jr, Chidsey CA, Pool PE, Braunwald E. Mechanism of norepinephrine depletion in experimental heart failure produced by aortic constriction in the guinea pig. Circ Res 1965;17:312-21.
- Böhm M, La Rosée K, Schwinger RHG, Erdmann E. Evidence of reduction of norepinephrine uptake sites in the failing human heart. J Am Coll Cardiol 1995;25:146-53.
- Blakely RD, De Felice LJ, Hartzell HC. Molecular physiology of norepinephrine and serotonin transporters. Trends Pharmacol Sci 1991;12:334-7.
- Galli A, Blakely RD, DeFelice LJ. Norepinephrine transporters have channel modes of conduction. Proc Natl Acad Sci USA 1996;93:8671-6.
- Fan THM, Liang CS, Kawashima S, Banerjee SP. Alterations in cardiac β-adrenergic receptor responsiveness and adenylate cyclase system by congestive heart failure in dogs. Eur J Pharmacol 1987;140:123-32.
- Bristow MR, Minobe W, Rasmusen R, Larrabee P, Skerl L, Klein JW, et al. Adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest 1992;89:803-15.
- Liang CS, Fan THM, Sullebarger JT, Sakamoto S. Decreased adrenergic neuronal uptake activity in experimental right heart failure: a chamber-specific contributor to beta-adrenoceptor down-regulation. J Clin Invest 1989;84:1267-75.
- Himura Y, Felten SY, Kashiki M, Lewandowski TJ, Delehanty JM, Liang CS. Cardiac noradrenergic nerve terminal abnormalities in dogs with experimental congestive heart failure. Circulation 1993;88:1299-309.
- Delehanty JM, Himura Y, Elam H, Hood WB Jr, Liang CS. β-Adrenoceptor downregulation in pacing-induced heart failure is associated with increased interstitial NE content. Am J Physiol Heart Circ Physiol 1994;266:H930-5.
- Kawai H, Mohan A, Hagen J, Dong E, Armstrong J, Stevens SY, et al. Alterations in cardiac adrenergic terminal function and β-adrenoceptor density in pacing-induced heart failure. Am J Physiol Heart Circ Physiol 2000;278:H1708-16.
- Leineweber K, Brandt K, Wludyka B, Beilflu B, Pönicke K, Heinroth-Hoffmann I, et al. Ventricular hypertrophy plus neurohumoral activation is necessary to alter the cardiac β-adreno-ceptor system in experimental heart failure. Circ Res 2002;91:1056-62.
- Leineweber K, Seyfarth T, Broddie OE. Chamber-specific alterations of noradrenaline uptake (uptake1) in right ventricles of monocrotaline-treated rats. Br J Pharmacol 2000;131:1438-44.
- Liang CS, Himura Y, Kashiki M, Stevens SY. Differential pre- and post-synaptic effects of desipramine on cardiac sympathetic nerve terminals in RHF. Am J Physiol Heart Circ Physiol 2002;283:H1863-72.
- Shite J, Dong E, Kawai H, Stevens SY, Liang CS. Selegiline improves cardiac noradrenergic nerve terminal function and β-adrenergic responsiveness in heart failure. Am J Physiol Heart Circ Physiol 2000;279:H1283-90.
- Dong E, Yatani A, Mohan A, Liang CS. Myocardial β-adreno-ceptor down-regulation by norepinephrine is linked to reduced norepinephrine uptake activity. Eur J Pharmacol 1999;384:17-24.
- Liang C, Rounds NK, Dong E, Stevens SY, Shite J, Qin F. Alterations by norepinephrine of cardiac sympathetic nerve terminal function and myocardial β-adrenergic receptor sensitivity in the ferret: normalization by antioxidant vitamins. Circulation 2000;102:96-103.
- Liang CS, Yatani A, Himura Y, Kashiki M, Stevens SY. Desipramine attenuates loss of cardiac sympathetic neurotransmitters produced by congestive heart failure and NE infusion. Am J Physiol Heart Circ Physiol 2003;284:H1729-36.
- Shite J, Qin F, Mao W, Kawai H, Stevens SY, Liang CS. Antioxidant vitamins attenuate oxidative stress and cardiac dysfunction in tachycardia-induced cardiomyopathy. J Am Coll Cardiol 2001;38:1734-40.
- Mao W, Iwai C, Qin F, Liang CS. Norepinephrine induces endoplasmic reticulum stress and downregulation of norepinephrine transporter density in PC12 cells via oxidative stress. Am J Physiol Heart Circ Physiol 2005;288:H2381-9.
- Mao W, Qin F, Iwai C, Vulapalli R, Keng PC, Liang CS. Extracellular norepinephrine reduces neuronal uptake of norepinephrine by oxidative stress in PC12 cells. Am J Physiol Heart Circ Physiol 2004;287:H29-39.
- Backs J, Haunstetter A, Gerber SH, Metz J, Borst MM, Strausser RH, et al. The neuronal norepinephrine transporter in experimental heart failure: evidence for a posttranscription down-regulation. J Mol Cell Cardiol 2001;33:461-72.
- Böhm M, Castellano M, Flesch M, Maach C, Moll M, Paul M, et al. Chamber-specific alterations of norepinephrine uptake site in cardiac hypertrophy. Hypertension 1998;32:831-7.
- Backs J, Bresch E, Lutz M, Kristen AV, Haass M. Endothelin-1 inhibits the neuronal norepinephrine transporter in hearts of male rats. Cardiovasc Res 2005;67:283-90.
- Moe GW, Albernaz A, Naik GO, Kirchengast M, Stewart DJ. Beneficial effects of long-term selective endothlin type A receptor blockade in canine experimental heart failure. Cardiovasc Res 1998;39:571-9.
- Yamauchi-Kohno R, Miyauchi T, Hoshino T, Kobayashi T, Aihara H, Sakai S, et al. Role of endothelin in deterioration of heart failure due to cardiomyopathy in hamsters. Increase in endothelin-1 production in the heart and beneficial effect of endothelin-A receptor antagonist on survival and cardiac function. Circulation 1999;99:2171-6.
- Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. For EARTH investigators. Long-term effects of darusentan on left-ventricular remodeling and clinical outcomes in the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet 2004;364:347-54.
- Kalra PR, Moon JC, Coates AJ. Do results of the ENABLE (Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol 2002;85:195-7.
- Patel AD, Iskandrian AE. MIBG imaging. J Nucl Cardiol 2002;9:75-94.
- Henderson EB, Kahn JK, Corbett JR, Jansen DE, Pippin JJ, Kulkarni P, et al. Abnormal I-123 metaiodobenzylguanidine myocardial washout and distribution may reflect myocardial adrenergic derangement in patients with congestive cardiomyopathy. Circulation 1988;78:1192-9.
- Ohshima S, Isobe S, Izawa H, Nanasato M, Ando A, Yamada A, et al. Cardiac sympathetic dysfunction correlates with abnormal myocardial contractile reserve in dilated cardiomyopathic patients. J Am Coll Cardiol 2005;46:2061-8.
- Ungerer M, Hartmann F, Karoglan M, Chlistalla A, Ziegler S, Richardt G, et al. Regional in vivo and in vitro characterization of autonomic innervation in cardiomyopathic human heart. Circulation 1998;97:174-80.
- Merlet P, Benvenuti C, Moyse D, Pouillart F, Dubois-Randé JL, Duval AM, et al. Prognostic value of MIBG imaging in idiopathic dilated cardiomyopathy. J Nucl Med 1999;40:917-23.
- Yamada T, Shimonagata T, Fukunami M, Kumagai K, Ogita H, Hirata A, et al. Comparison of the prognostic value of cardiac iodine-123 metaiodobenzylguanidine imaging and heart rate variability in patients with chronic heart failure: a prospective study. J Am Coll Cardiol 2003;41:231-8.
- Lotze U, Kaepplinger S, Kober A, Richartz BM, Gottschild D, Figulla HR. Recovery of the cardiac adrenergic nervous system after long-term β-blocker therapy in idiopathic dilated cardiomyopathy: assessment by increase in myocardial 123I-metaiodobenzylguanidine uptake. J Nucl Med 2001;42:49-54.
- de Milliano PAR, de Groot AC, Tijssen JGP, van Eck-Smit BLF, Van Zwieten PA, Lie KI. Beneficial effects of metoprolol on myocardial sympathetic function: evidence from a randomized, placebo-controlled study in patients with congestive heart failure. Am Heart J 2002;144:E3.
- Kasama S, Toyama T, Kumakura H, Takayama Y, Ichikawa S, Suzuki T, et al. Spironolactone improves cardiac sympathetic nerve activity and symptoms in patients with congestive heart failure. J Nucl Med 2002;43:1279-85.
- Kasama S, Toyama T, Kumakura H, Takayama Y, Ichikawa S, Suzuki T, et al. Effect of spironolactone on cardiac sympathetic nerve activity and left ventricular remodeling in patients with dilated cardiomyopathy. J Am Coll Cardiol 2003;41:474-81.
- Somsen GA, van Vlies B, de Miliano PA, Borm JJ, van Royen EA, Endert E, et al. Increased myocardial [123I]-metaiodobenzylguanidine uptake after enalapril treatment in patients with chronic heart failure. Heart 1996;76:218-22.
- Erol-Yilmaz A, Verberne HJ, Schrama TA, Hrudova J, De Winter RJ, Van Eck-Smit BLF, et al. Cardiac resynchronization induces favorable neurohumoral changes. Pacing Clin Electrophysiol 2005;28:304-10.
- The Beta-Blocker Evaluation of Survival Trial investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med 2001;344:1659-67.
- Eichhorn EJ, McGhie AL, Bedotto JB, Corbett JR, Malloy CR, Hatfield BA, et al. Effects of bucindolol on neurohormonal activation in congestive heart failure. Am J Cardiol 1991;67:67-73.
- Motherwell DW, Petrie MC, Martin W, Cobbe SM. 123I-metaiodo-benzylguanidine in chronic heart failure: is there a clinical use? Nucl Med Commun 2006;27:927-31.
- Bristow MR. β-Adrenergic receptor blockade in chronic heart failure. Circulation 2000;101:558-69.
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, et al. US Carvedilol Heart Failure Study Group. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med 1996;334:1349-55.
- The Cardiac Insufficiency Bisoprolol Study (CIBIS-II). a randomized trial. Lancet 1999;353:9-13.
- MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Chronic Heart Failure (MERIT-HF). Lancet 1999;353:2001-7.
- Ziegler D, Haxhiu MA, Kaan EC, Papp JG, Ernsberger P. Pharmacology of moxonidine: an I1-imidazoline receptor agonist. J Cardiovasc Pharmacol 1996;27 Suppl 3:S26-37.
- Dickstein K, Manhenke C, Aarsland T, Køff U, McNay J, Wiltse C. Acute hemodynamic and neurohumoral effects of moxonidine in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 1999;83:1638-44.
- Azevedo ER, Newton GE, Parker JD. Cardiac and systemic sympathetic activity in response to clonidine in human heart failure. J Am Coll Cardiol 1999;33:186-91.
- Cohn JN, Pfeffer MA, Rouleau J, Sharpe N, Swedberg K, Straub M, et al. MOXCON investigators. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). Eur J Heart Fail 2003;5:659-67.
- Kreusser MM, Haass M, Buss S, Hardt SE, Gerber SH, Kinscherf R, et al. Injection of nerve growth factor into stellate ganglia improves norepinephrine reuptake into failing hearts. Hypertension 2006;47:209-15.
- Münch G, Rosport K, Bültmann A, Baumgartner C, Li Z, Laacke L, et al. Cardiac overexpression of the norepinephrine transporter uptake-1 results in marked improvement of heart failure. Circ Res 2005;97:928-36.