
Novel squamosamide derivative (compound FLZ) attenuates Aβ25–35-induced toxicity in SH-SY5Y cells1
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive and irreversible memory loss due to neuronal death in aged people[1]. One of the major histopathological features of AD is the presence of senile plaques and concomitant neuronal loss in specific areas of the brain. β-Amyloid peptide (Aβ) is the main constituent of senile plaques and plays a critical role in the pathophysiology of AD[2–5]. Evidence has suggested that the neurotoxic effect of Aβ is related to the activation of the apoptosis pathway[6–9]. Neuronal apoptosis is the main cause of neuronal loss in patients with AD, thus, it has been proposed that neuroprotection may be a therapeutic strategy for slowing down the apoptotic process of related brain cells of AD patients[10].
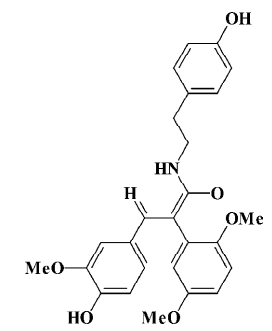
The natural squamosamide was isolated from the leaves of Annona squamosa. The compound FLZ is a novel synthetic cyclic derivative of natural squamosamide. Its chemical name is N-(2-[4-hydroxy-phenyl]-ethyl)-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxy-phenyl)-acrylamide and its molecular weight is 449.5 (Figure 1). Previous studies have demonstrated that FLZ has a potent neuroprotective property against experimental Parkinsonism and memory and learning deficits in mice[11,12]. In an in vitro study, FLZ was shown to protect against damage and apoptosis of primary cultured rat brain neurons, PC12, and SH-SY5Y cell lines exposed to hydrogen peroxide, glutamate, N-methyl-D-aspartate, dopamine, 1-methyl, 4-phenyl-pyridinium ion (MPP+), and ischemia-reoxygenation, indicating that FLZ possess a neuroprotective property[11,13]. Based on these results, the objective of this paper was to study the protective effect of FLZ on Aβ25–35-induced toxicity in human neuroblastoma SH-SY5Y cells and its active mechanism.
Materials and methods
Materials The compound FLZ was kindly provided by Professor Xiao-tian LIANG (Department of Pharmaceutical Chemistry, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China). It is a white powder with 99% purity. The compound was first dissolved in absolute ethanol and then diluted with 0.9% saline; the final ethanol concentration was <0.1% for use. Aβ25–35 (Sigma, St Louis, MO, USA) was dissolved in sterile double-distilled water at a concentration of 1 mmol/L stock solution, was aged at 37 °C for 4 d, and then stored at -20 °C before use. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), trypsin, 2',7'-dichlorofluoroescin diacetate (DCFH–DA), EDTA, propidium iodide (PI), rhodamine 123, agarose, HEPES, dithiothreitol (DTT), phenylmethylsul-fonyl fluoride (PMSF), Nonidet P40, and DNase were all purchased from Sigma (USA). Dulbecco’s Modified Eagle’s medium (DMEM), F-12, and fetal bovine serum (FBS) were obtained from Hyclone (Logan, UT, USA). The primary mouse monoclonal antibodies Bcl-2, Bax, and cytochrome c were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other regents were purchased from Beijing Chemical Company (Beijing, China).
SH-SY5Y cell cultures Human neuroblastoma SH-SY5Y cells were purchased from the Cell Center of the Institute of Basic Medical Science Research (Chinese Academy of Medical Sciences, China). The SH-SY5Y cells were cultured in DMEM:F12 (1:1) supplemented with 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified 95% oxygen and 5% CO2 atmosphere. The medium was changed every other day. The cells were cultured for 3–4 d until a confluence of 70%–80% was achieved. The trypan blue assay was used to count the cells and evaluate their viability as a percentage of viable and non-viable cells. The cell viability was >97% prior to the following experimental procedure. The cells were plated at an appropriate density according to each experimental protocol.
Treatment of SH-SY5Y cells On the day of the experiment, the medium was removed and the cells were then cultured in the same medium without FBS and exposed to the aged peptide Aβ25–35 for 48 h in the presence or absence of FLZ. Before adding Aβ25–35, different concentrations of FLZ (0.1, 1, and 10 µmol/L) were incubated with the cells for 30 min at 37 °C.
Cell viability assay After the cells were cultured with Aβ25–35 in the presence or absence of FLZ for 48 h, the cell viability was assayed with the MTT method[14]. Briefly, MTT was dissolved in the medium without FBS and added to cells grown in 96-well plates at a final concentration of 0.5 g/L. Following a 4-h incubation to allow its conversion into formazan crystals, the media was removed and cells were lysed with DMSO (Me2SO) to allow the crystals to dissolve. The absorbance was read at 570 nm using a Bio-Rad 450 microplate reader (Hercules, CA, USA). The results were expressed as a percentage of MTT reduction. The absorbance of the control cells (no FLZ and no Aβ25–35) was used as 100%.
Measurement of lactate dehydrogenase The medium was collected from the cells treated with Aβ25–35 for 48 h in the presence or absence of FLZ and centrifuged at 14 000×g for 5 min. The 50 µL of the supernatant was transferred to a tube, and the activity of lactate dehydrogenase (LDH) was determined using an LDH kit (Beijing Chemical Reagents Company, Beijing, China).
Apoptotic ratio assay by flow cytometry analysis Flow cytometry was used to assess the percentage of genomic DNA fragmentation in nuclei[15]. The cultured cells were trypsinized and harvested, centrifuged for 5 min at 200×g, washed twice with phosphate-buffered saline (PBS) and fixed in 70% alcohol overnight at 4 °C. The cells were centrifuged and washed with PBS, resuspended in 0.5 mL PBS containing 50 mg/L RNase A, and incubated for 1 h at 37 °C. The samples were stained with 50 mg/L PI for 30 min at 4 °C in the dark. The flow cytometry analysis was performed by FACScan (BD Biosciences, San Jose, CA, USA). Calculations of the percentage of apoptotic cells were based on the cumulative frequency curves of the appropriate DNA histograms. Debris was excluded from the collection of 10 000 nuclei by empirically setting the forward-scatter channel and side-scatter channel threshold levels.
Measurement of intracellular reactive oxygen species by flow cytometry analysis The production of intracellular reactive oxygen species (ROS) was determined using the fluorescent probe DCFH–DA[16]. DCFH–DA is a membrane permeable, non-fluorescent compound. In the presence of peroxides in cells, it is converted to the fluorescent derivative dichlorofluorescein (DCF). Following treatment with Aβ25–35 (25 µmol/L) for 48 h in the presence or absence of FLZ (0.1, 1, and 10 µmol/L), the cells were rinsed with PBS and incubated with DCFH–DA (20 µmol/L, final concen-tration) in Me2SO for 30 min at 37 °C. Loaded cells were washed 3 times, and the fluorescence intensity of DCF was determined by using flow cytometry (excitation=485 nm, emission=535 nm).
Measurement of intracellular glutathione The level of glutathione (GSH) was determined using o-phthalaldehyde (OPT)[17]. On the day of the experiment, the cultured medium was removed, the cells were washed 3 times with PBS, and lysed by vigorous shaking in the 500 µL buffer containing 0.2% Triton-X100 and 5 mmol/L EDTA (pH =8.3). After lysis, 0.3 mL of the buffer was removed and mixed with 0.1 mL 20% trichloric acetic acid, and centrifuged at 200×g for10 min. The supernatant was incubated with 50 mg/L OPT (dissolved in Me2SO and diluted to the final concentration with PBS) for 15 min at room temperature. The fluorescence was measured at an excitation wavelength of 350 nm and emission wavelength of 420 nm. The protein content was determined by the Lowry method[18].
Western blot analysis of cytochrome c, Bax, and Bcl-2 in SH-SY5Y cells Following the treatment of Aβ25–35, the cells were collected and washed with PBS. After centri-fugation, cell lysis was carried out at 4 °C by vigorous shaking for 30 min in RIPA buffer (25 mmol/L Tris-HCl, 150 mmol/L NaCl, 5 mmol/L EDTA, 5 mmol/L EGTA, 1 mmol/L PMSF, 1% TritonX-100, 0.5% Nonidet P40, 10 mg/L aprotinin, and 10 mg/L leupeptin). After centrifugation at 12 000×g for 15 min, the supernatant was separated and stored at -70 °C for the measurement of Bcl-2 and Bax[19].
The analysis of the cytochrome c release was performed as previously described[20]. The cells were collected and washed twice with PBS. After centrifugation, the cell pellets were suspended in 5 mL extraction buffer (in mmol/L: HEPES 50, KCl 50, EGTA 5, MgCl2 2, DTT 1, and PMSF 0.1, pH=7.4), and centrifuged after 15 min on ice. The cell pellets were resuspended in the above buffer, and homogenized in a Teflon-glass homogenizer (up and down, 50 strokes) after 45 min on ice. The homogenized buffer was transferred into the Eppendorf tube and centrifuged at 15 000×g for 20 min at 4 °C. The supernatants were removed and stored at -70 °C until use[20].
The protein concentration was determined by the Lowry method[18]. After the addition of the sample loading buffer, the protein samples (equal quantity) were denaturalized by heating at 100 °C and were separated on the 12% SDS–PAGE. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane were blocked for 1 h at room temperature in fresh blocking buffer (TBST, 0.1% Tween-20 in Tris-buffered saline, pH=7.4, containing 5% nonfat dried milk) and incubated with the primary antibody (dilution: Bcl-2 and Bax 1:200, cytochrome c 1:1000) for 3 h at room temperature. Following 3 washes with TBST, the membrane was incubated with alkaline phosphatase-conjugated secondary antibodies in TBST for 2 h at room temperature. The membrane was washed again 3 times in TBST buffer. The protein blot was visualized by 5-bromo-4-chloro-3'-indolyphosphate p-toluidine salt/nitro-blue tetrazolium chloride. The blot was scanned and analyzed the density using the software.
Statistical analysis All experiments were repeated at least 3 times using independent culture preparations. Quantitative data were expressed as mean±SD. The statistical analysis between various experimental results was performed using one-way ANOVA followed by the least significant difference test; P<0.05 was considered statistically significant.
Results
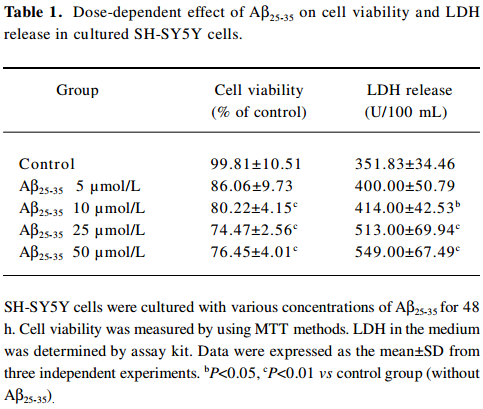
Effect of FLZ on cytotoxicity induced by Aβ25–35 in SH-SY5Y cells The exposure of SH-SY5Y cells to Aβ25–35 (10~50 µmol/L) for 48 h resulted in a significant decrease of cell viability and increase of LDH release into the medium in a dose-dependent manner (Table 1). The viability of the SH-SY5Y cells treated with Aβ25–35 (25 µmol/L) for 48 h was reduced to ~70% of that of the control (P<0.01), and the activity of LDH in the medium increased 1.5-fold (P<0.01). Most of the SH-SY5Y cellular morphology became round in shape and aggregated together (Figure 2). The 25 µmol/L Aβ25–35 was therefore used in the following study of the protective action of FLZ against Aβ25–35 neurotoxicity in SH-SY5Y cells.
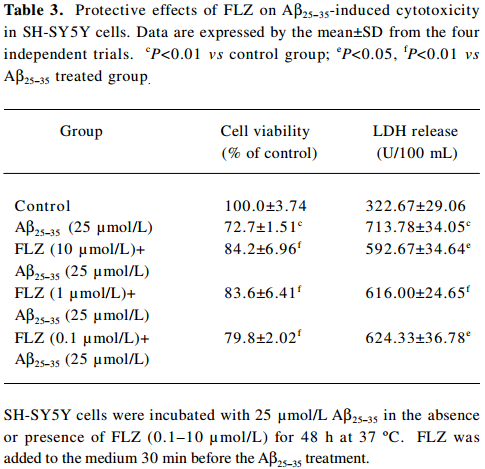
Full table
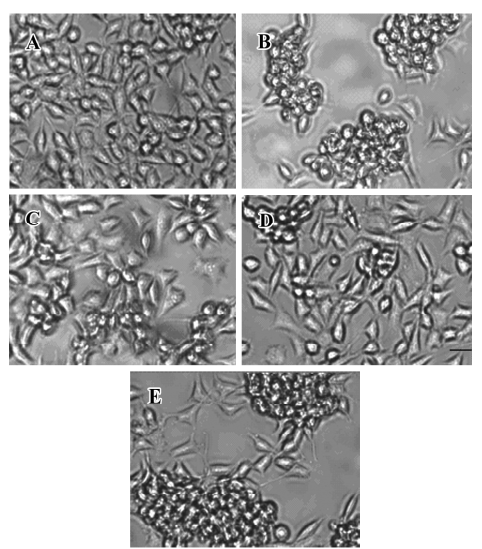
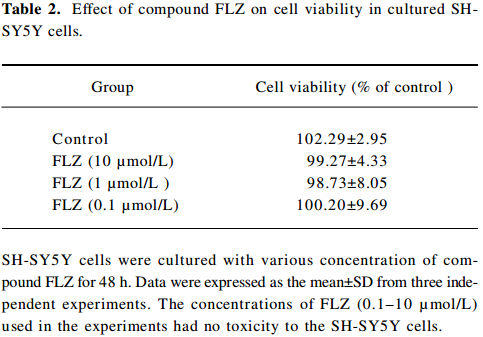
To select a non-cytotoxic concentration of FLZ in the study of the effect of FLZ on Aβ25–35 neurotoxicity, we first evaluated the effect of various concentrations (0.1, 1, and 10 µmol/L) of FLZ itself on the cell viability of SH-SY5Y cells. FLZ at concentrations of 0.1, 1, and 10 µmol/L showed no apparent cytotoxicity to SH-SY5Y cells (Table 2). Pretreatment of SH-SY5Y cells with 1 and 10 µmol/L FLZ for 30 min significantly protected the cells from Aβ25–35-induced cytotoxicity as demonstrated by increasing cell viability (P<0.01), and a concomitant reduction of LDH release (P<0.05) into the cultured medium (Table 3). The morphological injury of the SH-SY5Y cells were also reduced by FLZ treatment (Figure 2).
Full table
Full table
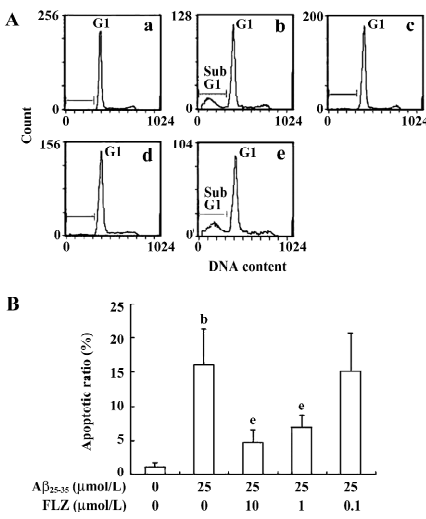
Effect of FLZ on apoptosis in SH-SY5Y cells induced by Aβ25–35 The hypodiploid sub-G1 peak is regarded as apoptotic cells. The results of the flow cytometry assay showed that the apoptotic ratio of the Aβ25–35-treated SH-SY5Y cells markedly increased as compared with that of untreated cells. FLZ (1 and 10 µmol/L) treatment significantly decreased the apoptotic cell accumulation in the sub-G1 peak in comparison with the Aβ25–35-treated SH-SY5Y cells, indicating that FLZ attenuated Aβ-induced apoptosis (Figure 3).
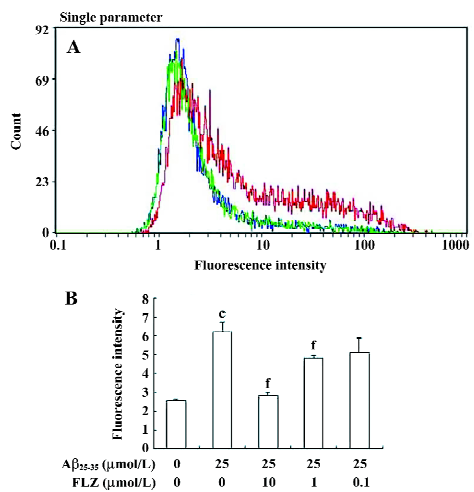
Effect of FLZ on ROS production and the GSH level induced by Aβ25–35 in SH-SY5Y cells Hydrogen peroxide was reported to mediate Aβ neurotoxicity. In our experiments, the exposure of SH-SY5Y cells to Aβ25–35 (25 µmol/L) induced a 2-fold increase in DCF fluorescence intensity, indicating that Aβ25–35 stimulated the production of ROS. The count of the high fluorescence intensity of the Aβ25–35-treated cells was also more than that of the untreated cells. The pretreatment of FLZ (10 µmol/L) almost completely inhibited the increase in DCF fluorescence in the SH-SY5Y cells (Figure 4).
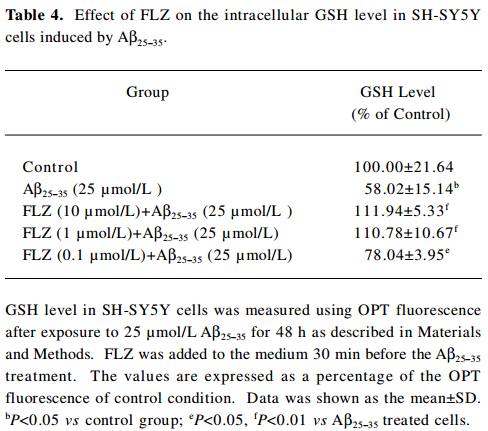
In contrast to the increase of ROS, the intracellular GSH level decreased dramatically in the SH-SY5Y cells after exposure to Aβ25–35 (25 µmol/L) for 48 h. The pretreatment of FLZ significantly attenuated the decrease of the GSH level induced by 25 µmol/L Aβ25–35 (Table 4).
Full table
Effect of FLZ on cytochrome c release, Bax, and Bcl-2 protein expressions in SH-SY5Y cells treated with Aβ25–35 The Western blot analysis showed that the treatment of SH-SY5Y cells with Aβ25–35 resulted in an increase of the pro-apoptotic Bax protein expression, while the expression of the anti-apoptosis Bcl-2 protein decreased. When the immunoblots were quantified by densitometry analysis, the ratio of Bax to Bcl-2 in the Aβ25–35-treated cells significantly increased as compared with the untreated cells. The pretreatment of SH-SY5Y cells with 1 and 10 µmol/L FLZ reversed the alternations of Bax and Bcl-2 expressions induced by Aβ25–35, and substantially reduced the ratio of Bax/Bcl-2 (Figure 5). The effect of FLZ at 0.1 µmol/L concentration based on the above criteria was weak.
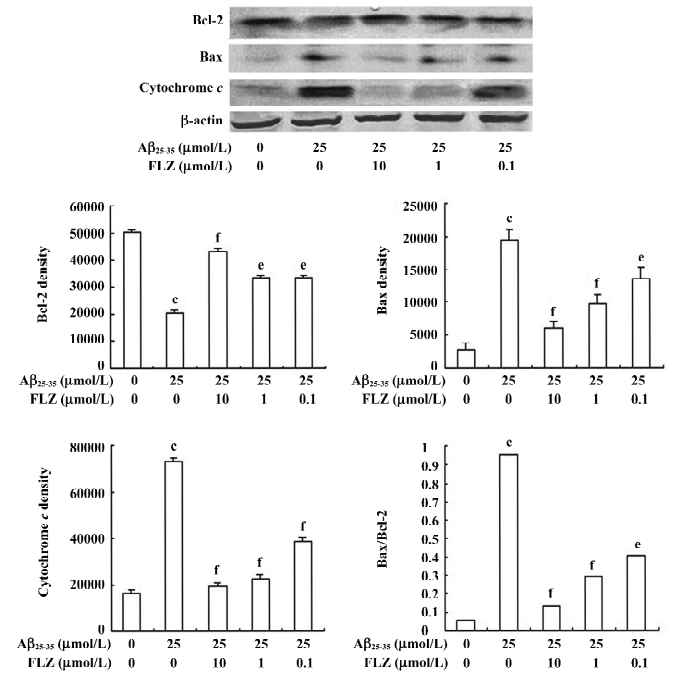
Aβ25–35 caused an increase of cytochrome c release from the mitochondria of SH-SY5Y cells. FLZ at concentrations of 1 and 10 µmol/L effectively blocked cytochrome c release from mitochondria of SH-SY5Y cells induced by Aβ25–35 (Figure 5).
Discussion
The accumulation of plaques containing Aβ in the brain is an invariant feature of AD pathology, and abundant evidence suggests that Aβ contributes to the etiology of AD[21]. Neuronal apoptosis was observed in human AD brains[22,23]. Several investigators reported that Aβ induced apoptosis in multiple cell types in vitro[19,24–26]. Aβ25–35 is considered to be the shorter toxic fragment exerting neurotoxic effects similar with Aβ1–40/42, such as learning and memory impairment, neuronal apoptosis, cholinergic dysfunction, and oxidative stress[27–29], so Aβ25–35 is usually used to establish the in vitro model of AD for the study of the neurotoxic properties of Aβ and for drug screening. The human dopaminergic neuroblastoma cell line SH-SY5Y is widely applied in different neurochemical research. Aβ was uptaken rapidly into the SH-SY5Y cells and reserved for several days[30]. Some evidence has been accumulated that suggests that Aβ25–35 induces neurotoxic effects in SH-SY5Y cells similar to the pathological changes of neurons in the mouse brain[24–26]. The results of the present paper also indicated that the exposure of SH-SY5Y cells to Aβ25–35 (25 µmol/L) for 48 h displayed remarkable injuries. The cell viability decreased and LDH release from the cells increased in a dose-dependent manner. Most of the cells became round in shape and aggregated together. The cell apoptotic ratio also increased significantly. When the SH-SY5Y cells were precultured with FLZ (1 and 10 µmol/L) for 30 min, all of the above Aβ25–35-induced injuries were significantly reduced. The cell viability, LDH release, and apoptotic ratio were all improved. The morphology of the FLZ-treated cells were close to that of the control cells. These results suggest that FLZ has protective action against Aβ25–35-induced neurocytotoxicity.
It is very important to study the mechanism by which FLZ exerts its protective action against Aβ25–-35-induced neurocytotoxicity. Although which signaling pathway mediated Aβ-induced neurotoxicity is not fully defined, oxidative stress has been proposed to play a key role[31–33]. Aβ stimulates the production of ROS by a direct or indirect pathway[31–34]. Several investigators have demonstrated that ROS is involved in the apoptotic mechanism of Aβ-mediated neurotoxicity and may contribute to the increase in the apoptotic processes found in AD[25,29,31–33]. The production of ROS can occur very early and cause damage to cardinal cellular components, such as lipid, protein, and nuclei acids, resulting in cell death by modes of apoptosis or necrosis. The high metabolic rate, a low concentration of GSH and the antioxidant enzyme catalase, and the large proportion of polyunsaturated fatty acids in the brain make brain tissue particularly vulnerable to oxidative damage[7]. Some studies have reported that free-radical scavengers or antioxidants, such as melatonin, EGb-761, vitamin E, and estrogen could attenuate the Aβ-induced apoptosis and neurocytotoxicity[25,29,31,35]. Some antioxidants were reported to be effective in the treatment of mild-to moderate dementia of AD patients[36,37]. It seems reasonable that antioxidants will play an important role in the search of drugs as pharmacotherapy of AD. Data from this present study showed that 25 µmol/L Aβ25–35 resulted in a significant increase of the ROS level in SH-SY5Y cells. This result is consistent with previous descriptions of the Aβ-mediated generation of ROS. In addition, the authors found that GSH, the most abundant antioxidant in cells, was depleted by the addition of Aβ25–35. The results suggested that oxidative stress was involved in Aβ-induced toxicity in SH-SY5Y cells.
As major sources of ROS, mitochondrial structures are exposed to high concentrations of ROS and might therefore be particularly susceptible to oxidative injury. It was reported that mitochondrial damage plays a pivotal role in cell apoptosis[7]. The present results show that Aβ25–35 induced mitochondrial dysfunction in the MTT analysis, because the MTT underwent conversion of the yellow MTT to purple formazan crystals by mitochondrial succinate dehydrogenase in viable cells, which primarily reflects the mitochondrial metabolic capacity of viable cells and the intracellular redox state[14]. Overproduction of ROS induced the opening of the mitochondrial permeability pore and caused the mitochondrial intermembrane space soluble protein (cytochrome c, apoptosis-inducing factor) release into the cytoplasm[38]. In most pathways of apoptosis, the release of mitochondrial cytochrome c and apoptosis-inducing factor are also key events in initiating the cascade of reactions leading to apoptotic cell death[39]. The release of cytochrome c is clearly regulated by the pro- and anti-apoptotic proteins of the Bcl-2 family (Bax, bak, bad, bim, and bid as pro-, and bcl-2 and bcl-xL as anti-apoptotic proteins). Bax promotes the release of cytochrome c from the mitochondria, and Bcl-2 inhibits the release of cytochrome c. The relative ratio of pro-apoptotic and anti-apoptotic proteins is important to determine cell survival or death[40–42]. The overexpression of Bcl-2 or Bcl-xL can inhibit free-radical generation and protects cells from apoptosis induced by various stimuli[43,44]. In the present study, Aβ25–35 treatment decreased the expression of Bcl-2, increased the expression of Bax, and promoted the cytochrome c release from mitochondria. The cell apoptotic ratio also significantly increased. In the previous study at our laboratory, Aβ25–35-induced cell apoptosis was also identified by other methods, such as the DNA ladder (data not shown).
In the present study, the pretreatment of FLZ significantly inhibited the increase of ROS generation and the decrease of GSH content in SH-SY5Y cells. In our previous study, FLZ was shown to inhibit microsomal lipid peroxidation induced by Fe2+-cysteine, and also to scavenge oxygen free radicals, indicating that FLZ has an antioxidant property[45]. Moreover, H2O2 is a donor of hydroxyl radical that has been reported to mediate Aβ protein toxicity[46]. The pretreatment of FLZ inhibited the H2O2-induced apoptosis of cells[11]. The pretreatment of FLZ also protected PC12/SH-SY5Y cells against dopamine/MPP+-induced apoptosis through inhibiting cytochrome c release and caspase 3 activation[11,13]. The present study further confirmed that FLZ reduced the Aβ-induced relative ratio of Bax and the Bcl-2 protein by increasing Bcl-2 and decreasing the Bax expressions; and also decreased the cell apoptotic ratio of SH-SY5Y cells. Taken together, it appears that the protective action of FLZ against Aβ25–35 neuron toxicity may be in part due to its antioxidant property.
There is debate as to whether treatment with antioxidants might theoretically act to prevent propagation of tissue damage and improve both survival and neurological outcomes[48]. The great pharmacological disadvantage of most antioxidants is their very limited passage through the blood–brain barrier. Therefore, antioxidants with much better blood–brain barrier permeability are required for their potential application in treating neurodegenerative diseases, such as AD[47]. Fortunately, the results of a pharmacokinetic study of FLZ in rats indicated that the oral administration of FLZ penetrates through the blood–brain barrier very well (data to be published).
Because apoptosis is the main cause in neurodegenera-tion, such as AD and Parkinson’s disease (PD), and oxidative stress is an early event in the apoptosis process, it appears that FLZ is a novel neuroprotectant to protect against oxidative injury and apoptosis, and a good candidate for neurodegeneration therapy. Further pharmacological and toxicological studies on FLZ are in progress.
References
- Famer D, Crisby M. Rosuvastatin reduces caspase-3 activity and up-regulates α-secretase in human neuroblastoma SH-SY5Y cells exposed to Aβ. Neurosci Lett 2004;371:209-14.
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K, et al. Amyloid plaque core protein in Alzheimer’s disease and Down syndrome. Proc Natl Acad Sci USA 1985;82:4245-9.
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992;256:184-5.
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem 1996; 271: 18 295–8.
- Butterfiled DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 2002;23:655-64.
- Wang R, Zhang HY, Tang XC. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by β-amyloid protein-(1-40) in rat. Eur J Pharmacol 2001;421:149-56.
- Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schussel K, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem Pharmacol 2003;66:1627-34.
- Xu J, Chen S, Ku G, Ahmed SH, Xu J, Chen H, et al. Amyloid β peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow and Metab 2001;21:702-10.
- Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA 1993;90:7951-5.
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995;267:1456-62.
- Feng W, Wei H, Liu G. Pharmacological study of the novel compound FLZ against experimental Parkinson’s models and its active mechanism. Mol Neurobiol 2005;31:295-300.
- Fang F, Liu GT. Protective effects of compound FLZ on β- amyloid peptide (25-35)-induced mouse hippocampal injury and learning and memory impairment. Acta Pharmacol Sin 2006;27:651-8.
- Zhang D, Zhang JJ, Liu GT. The novel squamosamide derivative (compound FLZ) attenuated 1-methyl, 4-phenyl-pyridinium ion MPP (+)-induced apoptosis and alternations of related signal transduction in SH-SY5Y cells. Neuropharmacology 2007;52:423-9.
- Zeng H, Chen Q, Zhao B. Genistein ameliorates β-amyloid peptide (25-35)-induced hippocampal neuronal apoptosis. Free Radic Biol Med 2004;36:180-8.
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991;139:271-9.
- Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 1983;130:1910-7.
- Hissin PJ, Hilf RA. Fluorimetric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 1976;74:214-26.
- Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265-75.
- Jang JH, Surh YJ. Protective effect of resvratrol on β-amyloid-induced oxidative PC12 cell death. Free Radic Biol Med 2003;34:1100-10.
- Takagi-Morishita Y, Yamada N, Sugihara A, Iwasaki T, Tsujimura T, Terada N. Mouse uterine epithelial aooptosis is associated with expression of mitochondrial voltage-dependent anion channels, release of cytochrome c from mitochondria, and the ratio of Bax to Bcl-2 or Bcl-XL. Biol Reprod 2003;68:1178-84.
- Bayer TA, Wirths O, Majtenyi K, Hartmann T, Multhaup G, Beyreuther K, et al. Key factors in Alzheimer’s disease: beta amyloid precursor protein processing, metabolism and intraneu-ronal transport. Brain Pathol 2001;11:1-11.
- Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol 1996;6:493-506.
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, et al. Activation of caspase 3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol 1999;155:1459-66.
- Watt JA, Pike CJ, Walencewicz-Wasserman AJ, Cotman CW. Ultrastructural analysis of β-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Res 1994;661:147-56.
- Feng Z, Zhang JT. Metatonin reduces amyloid β-induced apoptosis in pheochromeocytoma (PC12) cells. J Pineal Res 2004;37:257-66.
- Li YP, Bushnell AF, Lee CM, Perlmutter LS, Wong SK. Beta-amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res 1996;738:196-204.
- Olariu A, Tran MH, Yamada K, Mizuno M, Hefco V, Nabeshima T. Memory deficits and increased emotionality induced β-amyloid (25–35) are correlated with the reduced acetylcholine release and altered phorbol dibutyrate binding in the hippocampus. J Neural Transm 2001;108:1065-79.
- Tohda C, Tamura T, Konatsu K. Repair of amyloid β (25-35)-induced memory impairment and synaptic loss by a Kampo formula, Zokumei-to. Brain Res 2003;990:141-7.
- Bastianitto S, Ramassamy C, Dore S, Christen Y, Poirier J, Quirion R. The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by β-amyloid. Eur J Neurosci 2000;12:1882-90.
- Ida N, Masters CL, Beyreuther K. Rapid cellular uptake of Alzheimer amyloid beta A4 peptide by culture human neuroblastoma cells. FEBS Lett 1996;394:174-8.
- Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, et al. The role of oxidative stress in the toxicity induced by amyloid β-peptide in Alzheimer’s disease. Prog Neurobiol 2000;62:633-48.
- Behl C. Amyloid β-protein toxicity and oxidative stress in Alzheimer’s disease. Cell Tissue Res 1997;290:471-80.
- Peng QL, Buzzard AR, Lau BH. Neuroprotective effect of garlic compounds in amyloid β-peptide-induced apoptosis in vitro. Med Sci Monit 2002;8:BR328-37.
- Huang X, Atwood CS, Hartshom MA, Multhaup G, Goldstein LE, Scarpa RC, et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999;38:7609-16.
- Huang HM, Ou HC, Hsieh SJ. Antioxidants prevent amyloid peptide-induced apoptosis and alteration of calcium homeostasis in cultured cortical neurons. Life Sci 2000;66:1879-92.
- Maurizi CP. Loss of intraventricular fluid melatonin can explain the neuropathology of Alzheimer’s disease. Med Hypotheses 1997;49:153-8.
- Cherubini A, Martin A, Andres-Lacueva C, Di Iorio A, Lamponi M, Mecocci P, et al. Vitamine E level, cognitive impairment and dementia in older persons: the InCHIANTI study. Neurobiol Aging 2005;26:987-94.
- Skulachev VP. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett 1998;423:275-80.
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physicol Rev 2000;80:315-60.
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA 1998;95:4997-5002.
- Shacka JJ, Roth KA. Regulation of neuronal cell death and neurodegeneration by members of the Bcl-2 family: therapeutic implications. Curr Drug Targets CNS Neurol disord 2005;4:25-39.
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 2003;15:691-9.
- Jordan J, Galindo MF, Tornero D, Gonzalez-Gareia C, Cena V. Bcl-xL blocks mitochondrial multiple conductance channel activation and inhibits 6-OHDA-induced death in SH-SY5Y cells. J Neurochem 2004;89:124-33.
- Wiedau-Pazos M, Trudell JR, Altenbach C, Kane DJ, Hubbell WL, Bredesen DE. Expression of Bcl-2 inhibits cellular radical generation. Free Radic Res 1996;24:205-12.
- Xie P, Jiao XZ, Liang XT, Feng WH, Wei HL, Liu GT. Synthesis and antioxiactivity of squamosamide cyclic analogs. Acta Acad Med Sin 2004;26:372-8.
- Gilgun-Sherki Y, Melamed E, Offen D. Antioxidant treatment in Alzheimer’s disease: current state. J Mol Neurosci 2003;21:1-11.
- Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001;40:959-75.