
Resistin induces insulin resistance, but does not affect glucose output in rat-derived hepatocytes1
Introduction
Type 2 diabetes has been tightly linked to obesity by several lines of epidemiological and metabolic evidence[1]. A key site for lipid storage in mammals is adipose tissue, which can act as an endocrine and secretory organ by secreting a wide range of hormones and other protein factors called adipokines[2,3]. Among the latter is resistin, recently shown to be involved in insulin sensitivity and glucose tolerance[4,5]. Resistin was discovered while screening for genes that are downregulated in adipocytes by activators of the peroxisome proliferator-activated receptor nuclear receptor[4]. Resistin impaired insulin sensitivity in adipocytes and skeletal muscles by impairing insulin-mediated glucose transport[6–8]. Further studies show that administering resistin to rodents increases blood glucose levels[4,5]. Resistin knockout mice exhibit low blood glucose levels after fasting, due to reduced hepatic glucose production[9]. These findings support that the major effect of resistin is on the liver in the maintenance of blood glucose. However, the direct effect of resistin on hepatocytes is unknown.
Another protein involved in insulin regulation is the suppressor of cytokine signaling 3 (SOCS-3). It is a 32 kDa protein that belongs to a family of proteins (SOCS family) originally identified as inducible inhibitors of signal transduction by gp130 cytokines[10,11]. SOCS-3 inhibits insulin signal transduction by promoting the reduction tyrosine phosphorylation of insulin-induced insulin receptor substrate (IRS)[12]. Moreover, SOCS-3 may decrease insulin signal transduction by targeting IRS-1 and IRS-2 for proteasomic degradation[13].
In the present study, we found that resistin induced insulin resistance, but did not affect glucose output in H4IIE hepatocytes. We also investigated whether resistin and SOCS-3 interact to exert their effects on insulin signal transduction. We found that the effects of resistin on the insulin signaling pathway occurred at the level of IRS-1 and IRS-2 by inducing SOCS-3 and blocking SOCS-3; antisense oligodeoxynucleotides (ASO) prevented resistin from antagonizing insulin action. In this study, we investigated the direct action of resistin on rat-derived hepatocytes.
Materials and methods
Cell culture The rat hepatoma cell line H4IIE was cultured and maintained at 37 °C in an atmosphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Grand Island, NY, USA) containing 1 g/L glucose and 10% fetal bovine serum (Gibco BRL, Grand Island, NY, USA).
Viability tests The H4IIE hepatocytes were seeded at a density of 2×104 cells/well in 100 µL culture medium in 96-well microplates; a 3-(4,5-dimethyl-2-thiazol-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (Amersham, Picataway, UK) was performed. Before testing, the cells were incubated in serum-free DMEM (1 g/L glucose) overnight in the presence of recombinant rat resistin (100 ng/mL; Alexis, San Diego, CA, USA) for 8 h. Then they were incubated for 2 h at 37 °C in supplemented 0.5 g/L MTT, after which the formazan crystals were solubilized with DMSO. The optical density was recorded at 570 nm[14].
Glycogen detection The H4IIE hepatocytes were incubated in serum-free DMEM (1 g/L glucose) overnight before the assays were performed. The glucose levels were adjusted to 4.5 g/L and the H4IIE hepatocytes were treated with resistin (60 ng/mL). This dose was chosen because it is the physiological concentration of resistin as determined by our previous work that the serum levels of resistin are approximately 60 ng/mL in diet-induced, obese rats. To assess glycogen synthesis, the cells were lysed with 30% KOH and then incubated in vials at 100 °C for 20 min. After the addition of anhydrous ethanol, the vials were centrifuged at 4000×g for 15 min and the supernatants were discarded. Next, 0.5 mL distilled water and 1 mL of 0.2% anthrone (0.2 g of anthrone in 100 mL of 98% H2SO4 [g/mL], prepared freshly within 1 h) were added, and the vials were placed in boiling water for 20 min. The optical density at 620 nm of the solution in the vials was determined by photometry. This method could detect 1.6 µg glucose/mL, which was equivalent to 1.44 µg glycogen/mL[15].
Glucose output For the glycogenolysis analysis, the glucose output from H4IIE hepatocytes was determined during a subsequent 30 min, 1 h, 2 h, and 4 h incubation period in serum-free DMEM without glucose or pyruvate. Resistin (60 ng/mL) or 100 nmol/L glucagon (Sigma, St Louis, USA) was added. The medium was collected and stored at -20 °C until assayed for glucose using the glucose oxidase method (Rongsheng, Shanghai, China)[16].
Immunoblotting and immunoprecipitation The treated cells were collected, washed with ice-cold phosphate buffered saline, and lysed in protein lysis buffer (50 mmol/L Tris, 150 mmol/L NaCl, 10 mmol/L EDTA, 1% Triton X-100, 200 mmol/L NaF, and 4 mmol/L Na orthovanadate-containing protease inhibitors, pH 7.5) for 1 h on ice[17]. The protein concentration was measured by the Bradford method. IRS was immunoprecipitated from the cell lysates (200 µg) with 4 µg of antibody at 4 °C overnight. Protein A/G-agarose (Santa Cruz, CA, USA) was added to collect the immune complexes. The pellets were washed 3 times with lysis buffer, resuspended in Laemmli sample buffer and boiled for 90 s, and then centrifuged to save the supernatant prior to the SDS gels[4]. The proteins (10 µg/lane) were separated on 10% SDS–PAGE and transferred to nitrocellulose membranes.
The membranes were blocked with 5% bovine serum albumin (BSA) in TBST (50 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, and 0.05% Tween-20). The membrane was incubated in 2% BSA in TBST containing one of the following primary antibodies: IRS-1, IRS-2 (1:800; Cell Signaling, Danvers, MA, USA), anti-phospho-tyrosine (4G10, 1:800; Cell Signaling, Danvers, MA, USA), anti-phospho-Akt (Thr 308), or anti-phospho-Akt (Ser 473, 1:1000; Kangchen, Shanghai, China), anti-phospho-glycogen synthase kinase (GSK)-3β (Ser 9; 1:1000; Kangchen, Shanghai, China), SOCS-3 (1:500; Zhongshan, China), or GAPDH (1:2000; Kangchen, Shanghai, China). Next, the membranes were washed extensively with TBST, and then incubated with an appropriate secondary horseradish peroxidase-labeled antibody: antigoat, antimouse, or antirabbit (Santa Cruz, CA, USA) at a 1:4000 dilution. The bands were visualized by enhanced chemiluminescence (Amersham Biosciences, Picataway, UK).
RNA preparation and amplification by RT-PCR Total RNA was isolated from cultured H4IIE hepatocytes using the TRIzol method (Invitrogen, Madison, WI, USA). Single-strand cDNA synthesis was performed as follows: the reverse transcription mixture contained 1 µg total RNA, 0.5 µg of oligo d(T) primer, 4 µL of 5×RT buffer, 0.5 mmol/L deoxynucleotides, 50 U of RNase inhibitor, and 200 U of reverse transcriptase (Promega, Madison, WI, USA) in a total volume of 20 µL. The reaction was performed at 42 °C for 1 h followed by heat inactivation at 95 °C for 5 min. The number of cycles and reaction temperatures used in the PCR assay were optimized to provide a linear relationship between the amount of input template and the amount of PCR product. The primers used for the amplification, together with their specific optimum cycling conditions, were as follows:
Rat SOCS-3 (366 bp product): sense primer, 5'-CAC AGC AAG TTT CCC GCC GCC-3' and antisense primer, 5´-GTG CAC CAG CTT GAG TAC ACA-3', annealing temperature at 58 °C for 29 cycles.
β-actin (240 bp product): sense primer, 5'-TAA AGA CCT CTA TGC CAA CAC AGT-3' and antisense primer: 5'-CAC GAT GGA GGG GCC GGA CTC ATC-3' annealing temperature at 57°C for 25 cycles.
ASO against SOCS-3 mRNA The phosphorothioate-modified ASO sequences against SOCS-3 were as follows: sense, 5'-CAT GGT CAC CCA CAG-3' and antisense, 5'-CTG TGG GTG ACC ATG-3'[18] (Invitrogen, Shanghai, China). The H4IIE hepatocytes were transfected with the ASO at a final concentration of 500 nmol/L in serum-free DMEM and FuGENE6 (Roche, Basel, BS, Switzerland)[19].
Statistical analysis The data are presented as mean±SEM. The statistical analysis was performed using one-way ANOVA or the paired Student’s t-test where appropriate. Differences between groups were considered statistically significant when P<0.05.
Results
Resistin induces cellular insulin resistance in H4IIE hepatocytes Glycogen synthesis in insulin-stimulated hepatocytes is the most important marker of hepatocyte insulin sensitivity. After treatment with resistin for 2 h, basal glycogen synthesis decreased approximately 17% and insulin-stimulated glycogen synthesis decreased approximately 26% in the H4IIE cells (Figure 1).
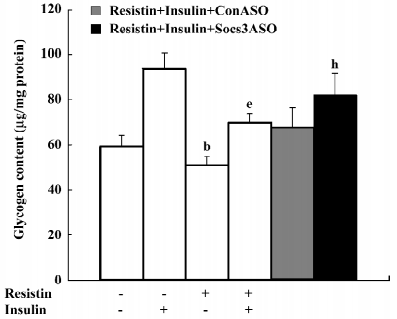
Importantly, cell viability was unaffected by resistin as assessed by the MTT assay. After treating the cells with 100 ng/mL resistin for 8 h, the optical density of the reaction product for the controls was 0.3067±0.018 and 0.3203±0.019 for the resistin-treated cells (n=12).
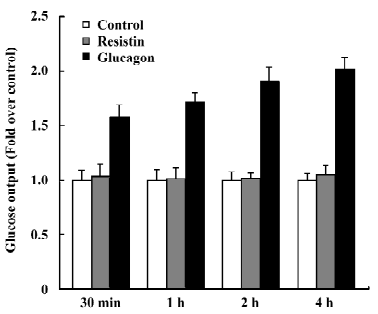
Resistin does not affect glucose output After incubation with 60 ng/mL resistin for 30 min, 1 h, 2 h, and 4 h, glucose output was unchanged compared to the control (Figure 2). In this experiment, glucose output after stimulation with 100 nmol/L glucagon was used as a positive control.
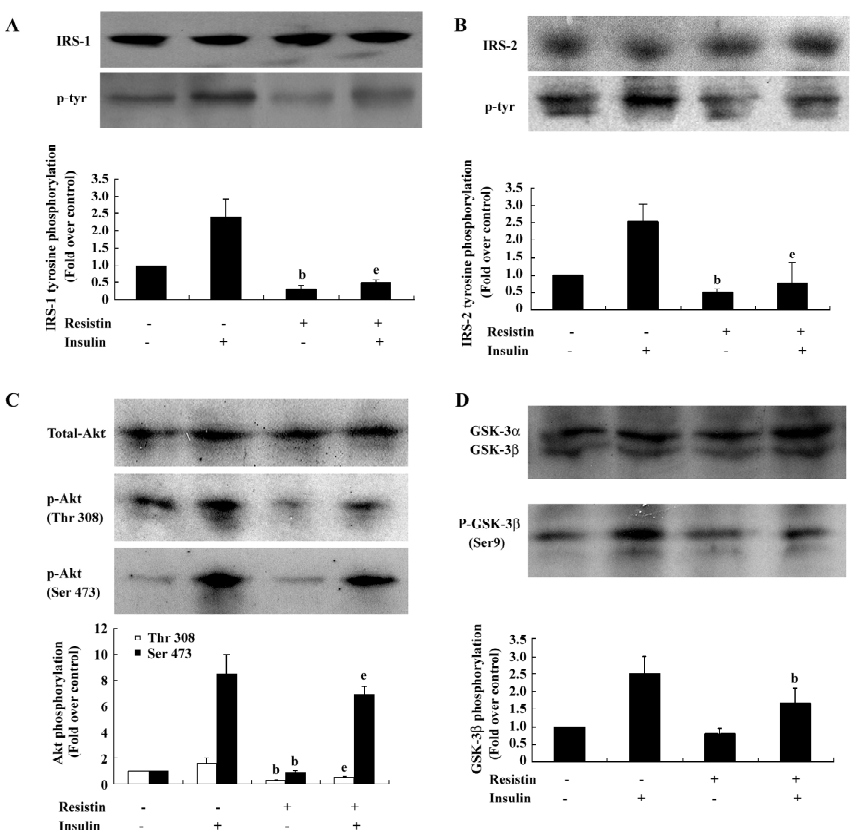
Resistin inhibits insulin-stimulated IRS-1, IRS-2, Akt, and GSK-3β phosphorylation To determine the mechanisms contributing to resistin-induced insulin resistance, we investigated whether resistin affects the IRS/PI3K/PDK/Akt/GSK pathway, which is stimulated by insulin during glycogen synthesis[20]. Resistin decreased insulin-stimulated tyrosine phosphorylation of IRS-1 by 79% without affecting the total protein levels of IRS-1 (Figure 3A). In parallel, the insulin-stimulated tyrosine phosphorylation of IRS-2 decreased by approximately 70% by resistin (Figure 3B). IRS-associated Akt phosphorylation was reduced in the resistin-treated hepatocytes (Figure 3C). Furthermore, the downstream activation of GSK by insulin was diminished by 33% by resistin (Figure 3D). These results showed that resistin inhibited the insulin-stimulated phosphorylation of IRS-1, IRS-2, Akt, and GSK-3β, but did not significantly affect the total protein levels. Thus, resistin induced cellular insulin resistance in rat-derived hepatocytes at the steps of IRS phosphorylation.
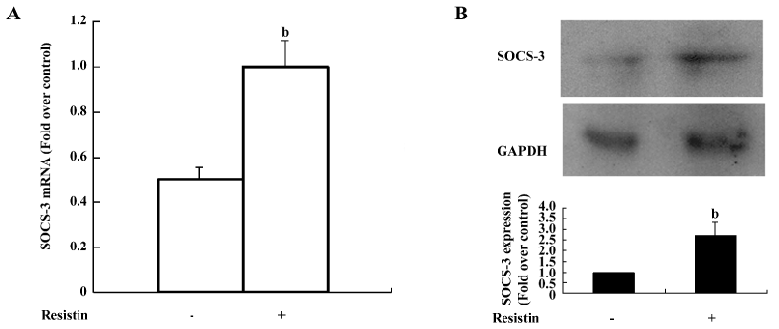
Resistin upregulates SOCS-3 expression SOCS-3 negatively regulates insulin signaling by 2 distinct mechanisms. First, SOCS-3 promotes the reduction of insulin-induced IRS tyrosine phosphorylation[12]. Second, SOCS-3 targets IRS-1 and IRS-2 for proteasomic degradation[13]. Since we found that resistin also decreased IRS phosphorylation, we aimed to determine whether resistin exerts its effects on insulin signal transduction via modulating SOCS-3. SOCS-3 mRNA was significantly increased 2-fold over the control (Figure 4A). The SOCS-3 protein level was upregulated approximately 2.7-fold by resistin (60 ng/mL for 2 h) compared with the control (Figure 4B).
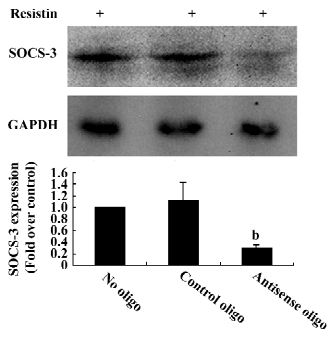
Blocking SOCS-3 impairs resistin antagonism of insulin action The sequence 5´-CTG TGG GTG ACC ATG-3´ was capable of reducing 70% resistin-stimulated SOCS-3 expression (Figure 5). Resistin decreased both basal and insulin-stimulated glycogen synthesis. Basal glycogen synthesis decreased by approximately 17% and insulin-stimulated glycogen synthesis was reduced by approximately 26% in cells treated for 2 h with resistin (Figure 1). This reduced insulin-stimulated glycogen synthesis by resistin was decreased by 50% when SOCS-3 was inhibited via ASO (Figure 1). Thus, the effect of resistin on insulin signal transduction may be mediated in part through its regulation of SOCS-3.
Discussion
In this study we examined the direct effect of resistin on glucose output and glycogen synthesis and metabolism in rat hepatocyte cells. We show that resistin induced cellular insulin resistance, but did not affect glucose output in hepatocytes. We have shown that resistin inhibits basal and the insulin-stimulated phosphorylation/activation of IRS-1and IRS-2, and its downstream targets Akt and GSK. SOCS-3 may decrease IRS-1 and IRS-2 phosphorylation[12,13]. SOCS-3 was found to be a cellular mediator of the ability of resistin to antagonize insulin action in adipocytes[8]. We show that the resistin-induced SOCS-3 expression also appears to mediate resistin’s inhibitory effects on IRS, Akt, and GSK in rat-derived hepatocytes.
Resistin plays a role in diet-induced hepatic insulin resistance[21]; resistin knockout mice exhibit low blood glucose levels after fasting[9]. These findings support a physiological function for resistin in the maintenance of blood glucose. The overexpression of resistin impairs the glucose tolerance in adult human hepatocytes[22], but serum and plasma resistin levels were either reduced or increased in type 2 diabetes patients with no significant correlation with homoeostasis model assessment for insulin resistance, waist circumference, body mass index, or total cholesterol[23]. These studies suggest that resistin is unlikely to play a critical endocrine role in insulin resistance or energy homoeostasis in humans. Studies have found that resistin participates in the regulation of fasted blood glucose in resistin knockout mice[9]. Administering resistin to rodents increases blood glucose levels[4,5]. These findings suggest that the liver is another important resistin target organ. So it is necessary to investigate the function of resistin on rodent hepatocytes.
Resistin affects insulin regulation in several different cell types. In L6 rat skeletal muscle cells, it does not alter insulin receptor signaling, but does affect insulin-stimulated glucose uptake, presumably by decreasing the intrinsic activity of cell surface glucose transporters[6]. Insulin-stimulated glucose uptake is also inhibited by resistin in matured 3T3-L1 adipocytes[8].
In this study we examined the direct effect of resistin on glycogen synthesis, glucose output, and the metabolism in H4IIE hepatocytes. First, we demonstrated that resistin inhibits both basal and insulin-stimulated glycogen synthesis, but does not affect glucose output in H4IIE hepatocytes. In hyperinsulinemic states, the hepatic glucose output was upregulated in resistin-like molecule β transgenic mice, but the hepatic glucose output was unchanged in normal states in resistin-like molecule β transgenic mice[24]. Resistin affects glucose metabolism and may be insulin-dependent, similar to resistin-like molecule β. Insulin clamps studies have demonstrated that resistin infusion increased hepatic glucose output[25]. In that case, the serum resistin concentration is far more than our 60 ng/mL, which is the physiological concentration of resistin in diet-induced obese rats. High concentration of resistin, not the physiological concentration may increase glucose output.
Next, we sought to elucidate the mechanisms underlying the resistin-induced impairment of glycogen synthesis by studying the IRS/Akt/GSK pathway. Resistin affected the insulin signal pathway at the level of IRS-1 and IRS-2, as evidenced by the decreased insulin-dependent phosphorylation of IRS and comparable reductions in downstream signals, including the phosphorylation of Akt and GSK.
Resistin was also shown to increase SOCS-3 mRNA and protein in H4IIE hepatocytes. SOCS-3 belongs to the SOCS protein family, which is composed of 8 members, all possessing a common structure that displays a variable N-terminal region, a central SH2 domain, and a C-terminal tail, named the SOCS box motif[26]. SOCS-3 binds to phosphorylated insulin receptors and competitively interferes with the binding of other SH2 domain-containing proteins[26]. The induction of SOCS-3 in the liver is an important mechanism for interleukin-6-mediated insulin resistance[27]. Blocking SOCS-3 expression by ASO showed that the loss of SOCS-3 function impairs the resistin antagonism of insulin action. Thus, the induction of SOCS-3 in H4IIE hepatocytes also appears to be an important mechanism by which resistin induces insulin resistance.
In summary, we found that the function of resistin on the liver was to induce insulin resistance, but not to impair glucose output. Resistin attenuated multiple effects of insulin, including insulin-stimulated glycogen synthesis and the phosphorylation of IRS, protein kinase B/Akt, as well as GSK-3β. Furthermore, resistin may antagonize insulin action by increasing SOCS-3 at both the mRNA and protein levels. Taken together, these results show that the induction of SOCS-3 is an important mechanism by which resistin induces insulin resistance.
References
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000;106:473-81.
- Hansen HO, Andreasen PH, Mandrup S, Kristiansen K, Knudsen J. Induction of lipogenesis during differentiation in a “preadipocyte” cell line. J Biol Chem 1976;251:6462-4.
- Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab 2002;13:18-23.
- Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature 2001;409:307-12.
- Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA 2001;98:502-6.
- Palanivel R, Maida A, Liu Y, Sweeney G. Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia 2006;49:183-90.
- Fan HQ, Gu N, Liu F, Fei L, Pan XQ, Guo M, et al. Prolonged exposure to resistin inhibits glucose uptake in rat skeletal muscles. Acta Pharmacol Sin 2007;28:410-6.
- Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol 2005;25:1569-75.
- Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, et al. Regulation of fasted blood glucose by resistin. Science 2004;303:1195-8.
- Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997;387:921-4.
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997;387:917-21.
- Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, . SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-α in the adipose tissue of obese mice. J Biol Chem 2001; 276: 47 944–9.
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 2002; 277: 42 394–8.
- Janjic D, Wollheim CB. Islet cell metabolism is reflected by the MTT (tetrazolium) colorimetric assay. Diabetologia 1992;35:482-5.
- Chun Y, Yin ZD. Glycogen assay for diagnosis of female genital Chlamydia trachomatis infection. J Clin Microbiol 1998;36:1081-2.
- Gauna C, Delhanty PJ, Hofland LJ, Janssen JA, Broglio F, Ross RJ, et al. Ghrelin stimulates, whereas des-octanoyl ghrelin inhibits, glucose output by primary hepatocytes. J Clin Endocrinol Metab 2005;90:1055-60.
- Staubs PA, Nelson JG, Reichart DR, Olefsky JM. Platelet-derived growth factor inhibits insulin stimulation of insulin receptor substrate-1-associated phosphatidylinositol 3-kinase in 3T3-L1 adipocytes without affecting glucose transport. J Biol Chem 1998; 273: 25 139–47.
- Butler M, McKay RA, Popoff IJ, Gaarde WA, Witchell D, Murray SF, et al. Specific inhibition of PTEN expression reverses hyperglycemia in diabetic mice. Diabetes 2002;51:1028-34.
- Calegari VC, Bezerra RM, Torsoni MA, Torsoni AS, Franchini KG, Saad MJ, et al. Suppressor of cytokine signaling 3 is induced by angiotensin II in heart and isolated cardiomyocytes, and participates in desensitization. Endocrinology 2003;144:4586-96.
- Valverde AM, Burks DJ, Fabregat I, Fisher TL, Carretero J, White MF, et al. Molecular mechanisms of insulin resistance in IRS-2-deficient hepatocytes. Diabetes 2003;52:2239-48.
- Muse ED, Obici S, Bhanot S, Monia BP, McKay RA, Rajala MW, et al. Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest 2004;114:232-9.
- Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J 1995;14:2816-26.
- Zhou L, Li Y, Xia T, Feng S, Chen X, Yang Z. Resistin overexpression impaired glucose tolerance in hepatocytes. Eur Cytokine Netw 2006;17:189-95.
- Kushiyama A, Shojima N, Ogihara T, Inukai K, Sakoda H, Fujishiro M, . Resistin-like molecule beta activates MAPKs, suppresses insulin signaling in hepatocytes, and induces diabetes, hyperlipidemia, and fatty liver in transgenic mice on a high fat diet. J Biol Chem 2005; 280: 42 016–25.
- Youn BS, Yu KY, Park HJ, Lee NS, Min SS, Youn MY, et al. Plasma resistin concentrations measured by enzyme-linked immunosorbent assay using a newly developed monoclonal antibody are elevated in individuals with type 2 diabetes mellitus. J Clin Endocrinol Metab 2004;89:150-6.
- Rajala MW, Obici S, Scherer PE, Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 2003;111:225-30.
- Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 2003;278:13740-6.