
Early lung injury contributes to lung fibrosis via AT1 receptor in rats
Introduction
Pulmonary fibrosis (PF) is a progressive and often fatal human disease characterized by irreversible fibrosis resulting from excessive repair of damaged alveoli[1]. While early studies have suggested that fibrosis occurs in the interstitium of the lungs, more recent studies have suggested that the initial injury in alveolar epithelial cells (AEC) causes intra-alveolar fibrosis and the collapse of alveolar capillary units[2]. These latter observations suggest that PF may be an ongoing form of acute lung injury occurring sequentially at discrete sites in the lungs. The evolving theory about the pathogenesis of PF is that severity of fibrogenic response in the lungs is directly related to the severity of lung injury in the early phase, including sequential AEC injury, capillary endothelial cells injury, and infiltrations of inflammatory cells[3]. The observations emphasize that effective therapies for these disorders must be given early in the natural history of the disease, prior to the development of extensive lung destruction and fibrosis.
Bleomycin is an anticancer agent prescribed for various cancers, including that of the lungs. However, this drug has a dose-dependent pulmonary toxicity, including lung fibro-sis, which limits its clinical use[4]. The toxic effect of this agent has been utilized advantageously in a number of experimental approaches to induce PF in animal models. Several groups of workers have reported that in the early stages of bleomycin-induced lung damage, the lesions are associated with biochemical and functional changes that resemble those of human PF, including inflammatory cell infiltration, increased collagen content, and reduced lung volume and compliance[5,6]. It is useful to assess the effects of potential therapeutic agents.
Angiotensin II, which is mainly generated by the angiotensin converting enzyme (ACE) and chymase, is a peptide that plays a crucial role in regulating blood pressure and sodium homeostasis[7]. Recent studies have shown that angiotensin II is also closely associated with tissue injury and fibrogenesis in circulatory organs[8] and lungs[9]. The vast majority of the actions of angiotensin II are thought to be mediated via the angiotensin type I (AT1) receptor. The AT1 receptor has been localized to fibroblasts, vascular smooth muscle cells, macrophages, and AEC in patients with PF induced by chronic obstructive pulmonary disease[10]. Human lung fibroblast proliferation and the synthesis of the extracellular matrix in human lung fibroblasts increased after stimulation by angiotensin II in vivo[11]. The AT1 receptor mediates AEC apoptosis in response to angiotensin II[12]. A high concentration of ACE has been observed in bronchoalveolar lavage (BAL) fluid from patients with idiopathic pulmonary fibrosis (IPF)[13]. In an animal model of radiation-induced PF, concentrations of ACE and angiotensin II increased in lung tissue homogenates[14]. The administration of an ACE inhibitor or an AT1 receptor antagonist significantly attenuated PF in animal models[15,16]. These studies suggest that the process of PF is promoted by an activated local renin-angiotensin system in the lungs via the AT1 receptor. Taken together, the evidence from these studies suggest that angiotensin II plays an important role in the promotion of PF, and the AT1 receptor is a prominent receptor for the transmission of its signaling. However, these studies focused on fibrogenic progression, and the relationship between the AT1 receptor and lung injury in the early phase in vivo remains unclear. The aims of this study are to clarify whether the AT1 receptor located in the lungs is induced in the early stage of PF induced by bleomycin, and whether early lung injury could be reduced by valsartan, an antagonist of the AT1 receptor.
Materials and methods
Animals Male Sprague-Dawley (SD) rats (200–220 g; Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China) were maintained in a controlled environment and provided with water and standard rodent food. All rats were acclimatized to their new surroundings for 1 week prior to the animal experiments, which were performed at the Animal Department (Shanghai Institute of Material Medica, Shanghai, China) and approved by the Shanghai Animal Care and Use Committee.
Experimental protocols One hundred and ninety SD rats were randomly divided into 7 groups (n=40 in A, B, C, D groups, n=10 in E, F, G groups). The rats in group A and B were injected intratracheally with 2 mL/kg saline, and then given saline or 60 mg·kg-1·d-1 valsartan gavaged at 9:00 AM for 21 d. Those in group C, D, E, F and G were given saline, valsartan (60, 30, 15 mg·kg-1·d-1, Qianhong, Changzhou, China), or 0.5 mg·kg-1·d-1 dexamethasone (Taihe, Tianjin, China) gavage at 9:00 AM for 21 d after an intratracheal injection of bleomycin sulfate (8 U/kg, 16 U/mL in saline, Sigma, St Louis, MO, USA). The day of intratracheal injection with bleomycin or saline was designated d 0. The rats were eutha-nized by sodium barbital (80 mg/kg, intraperitoneal) on d 1, 3, 7 and 21 after bleomycin or saline administration. The lung vasculature was perfused free of blood, and then the left lungs were removed from the trachea and hilar nodes and weighed. The left lungs of half of the animals were fixed in 4% phosphate buffered paraformaldehyde for histopathological preparation. BAL fluid samples were collected from the right lung of each animal (5 rats from each group) for inflammatory cell count and protein concentration assay. For all the animals, recovery of lavage fluid was about 80% of the utilized lavage volume. Left lung tissues were frozen in liquid nitrogen for measurements of hydroxyproline, malondialdehyde, myeloperoxidase, ACE activity, caspase-3 activity, mRNA expression, and protein expression profiles.
Histopathological evaluation Rat lung tissues were processed for routine paraffin embedding, and serial sections (5 μm) were stained with hematoxylin and eosin (H&E) and a modified Masson trichrome to assess the degree of inflammation and fibrosis. Fibrosis and alveolitis were scored by blinding using the previously described semiquantitative criteria[17].
Hydroxyproline assay in lung tissues Frozen lung tissues were homogenized by a Polytron tissue homogenizer in saline containing 0.1 mol/L phenylmethylsulfomyl fluoride. The homogenized sample was hydrolyzed in 6 mol/L HCl and the hydroxyproline concentration was quantified by chloramine-T in duplicate lung tissue samples as previously described[18].
Malondialdehyde concentration, ACE activity, and myeloperoxidase activity assay in lung tissues Lung tissue samples were homogenized at a w/v ratio of 1:10 in cold Tris-HCl buffered saline (pH 7.4, 10 mmol/L Tris-HCl, 0.1 mmol/L EDTA-2Na, 10 mmol/L Saccharose, 0.8% sodium chloride solution). One portion of each homogenate was centrifuged at 3000×g for 10 min at 4 °C, and the supernatant was used for the measurement of malondialdehyde concentration and ACE activity. The malondialdehyde concentration was determined by the thiobarbituric acid method as previously described[19]. ACE activity was analyzed, as previously described[20], by measuring the hydrolysis of Hippuryl-L-histidyl-L-leucine (Hip-His-Leu; Sigma, USA) substrate to histidyl-leucine (His-Leu), which fluoresces at an excitation wavelength of 365 nm and an emission wavelength of 495 nm. The reaction was stopped by 0.28 mol/L NaOH. o-Phthalaldehyde solution (Sigma, USA) bound to the His-Leu product, and the amount of tagged His-Leu was determined by spectrofluorophotometry (RF-5301 PC spectro-fluorophotometer; Shimadzu, Kyoto, Japan). Another portion of each homogenate was used to analyze myeloperoxidase activity with a microplate reader (Molecular Devices, Sunnyvale, CA, USA) using a myeloperoxidase activity kit (Jiancheng, Nanjing, China). Changes in absorbance at 450 nm were measured with the microplate reader.
Caspase-3 activity assay in lung tissues Caspase-3 activity was determined according to the method of Stennicke and Salvesen[21]. Lung tissue samples were suspended at a w/v ratio of 1:10 in Tris-HCl buffered saline, and homogenized 3 times at 4 °C. The homogenate was centrifuged at 12 000×g for 10 min at 4 °C, and pre-incubation was carried out in a medium containing 150 µg protein of the cytosolic sample and 1 mL caspase buffer {20 mmol/L Pipes, 100 mmol/L NaCl, 10 mmol/L dithiothreitol, 1 mmol/L EDTA, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), and 10% sucrose, pH 7.2} at 37 °C for 0.5 h, followed by incubation with 10 µL fluorogenic substrate Ac-DEVD-AMC (N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-methyl-coumarin; Sigma, USA). Active caspase-3 cleaved the substrate and released the fluorogenic AMC. AMC fluorescence was quantified by spectrofluorophotom-etry using an excitation wavelength of 380 nm and an emission wavelength of 460 nm. For the control, we added 10 µL of Ac-DEVD-CHO (N-Acetyl-Asp-Glu-Val-Asp-al; Sigma, USA), the specific inhibitor blocking cleavage of the fluorogenic substrate, to the incubation medium.
Protein and cell count in BAL fluid The total cell number in BAL fluid were examined immediately following lavage. The remainder of each sample was centrifuged at 1200×g for 15 min at 4 °C. The supernatant fractions were used to measure total protein concentration. The protein concentration in BAL fluid was measured using the bicinchoninic acid (BCA) method (Pierce Chemical, Rockfold, IL, USA) and expressed as grams of protein per liter of BAL fluid (g/L).
Collagen I and collagen III gene expression by RT-PCR Lung tissues were ground into a powder in liquid nitrogen, and the gene expression of collagen I, collagen III, and GAPDH were measured using RT-PCR. Total RNA was extracted using the TRIzol Reagent (Invitrogen Life Techno-logies, Paisley, UK) according to the manufacturer’s instructions. The yield and purity of the isolated RNA solution were determined by A260/A280 readings on a spectro-photometer. Reverse transcription was performed on 2 µg of RNA with random primers and Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA).The PCR were carried out with the primers shown in Table 1. Amplifications consisted of 10 min at 95 °C, following 30 cycles of 60 s at 95 °C, 45 s at 49–62 °C, and 75 s at 72 °C. The PCR products were analyzed by electrophoresis on an agarose gel, stained with ethidium bromide, and photographed.
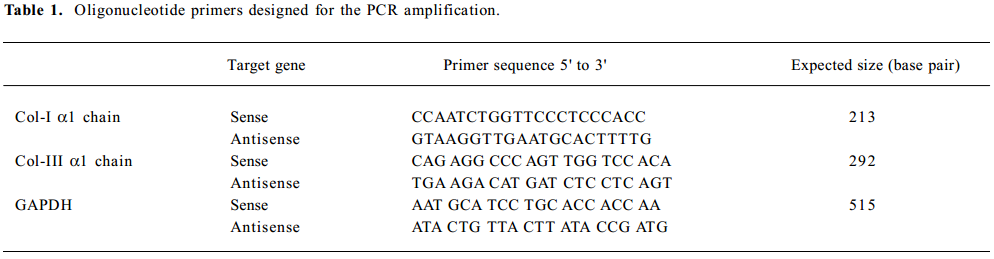
Full table
Western blot analysis of AT1 Supernatants of the lung tissue homogenate were collected after 30 min of centrifugation at 12 000 ×g, and protein concentration was measured by BCA protein assay (Pierce Chemical, USA). An aliquot of 20 µg supernatants were placed in sample buffer (4% sodium dodecyl sulfate, 125 mmol/L Tris-HCl, pH 6.8, 1% β-mercaptoethanol, 50% (v/v) glycerol, and 0.01% (w/v) bromophenol blue) and denatured at 100 °C for 10 min. The denatured samples were separated on 8% sodium dodecyl sulfate-polyacrylamide electrophoresis gels. The proteins were transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Bucks, UK) by electroblotting. The blots were blocked for 1 h with 5% (w/v) fat-free milk powder in phosphate buffered solution (PBS) (0.1% Tween-20), and subsequently incubated with primary rabbit antibodies against the AT1 receptor (Novus Biologicals, Littleton, CO, USA) and mouse primary antibodies against GAPDH (Abcam, Cambridge, UK) at 4 °C overnight. The blots were washed 3 times in PBS with 0.1% Tween-20. They were then incubated for 1 h with anti-rabbit (Abcam, UK) and anti-mouse (Abcam, UK) antibodies conjugated with horseradish peroxidase. Immunoreactive bands were detected by chemiluminescence (Amersham Pharmacia Biotech, UK) and exposure to Kodak film (Eastman Kodak, USA).
In situ detection apoptosis Briefly, paraffin-embedded tissues were sectioned (5 µm), and antigen retrieval was performed using citrate buffer. In situ detection of DNA fragmentation was made by TUNEL assay (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The slides were then observed under a fluorescence microscope (Leica, Mannheim, Germany).
Statistical methods Results are given as mean±SD. One-way ANOVA followed by the least significant difference method was used to determine differences among groups for all continuous parameters. The Mann-Whitney or Chi-square tests were applied for non-continuous parameters. The statistical significance level was set at P<0.05.
Results
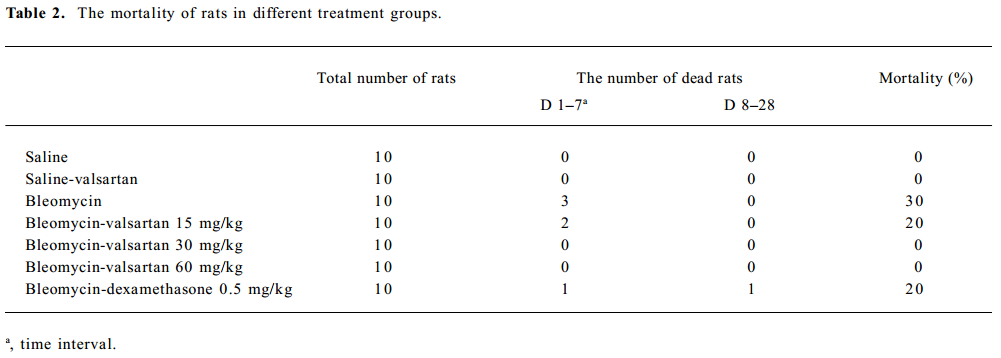
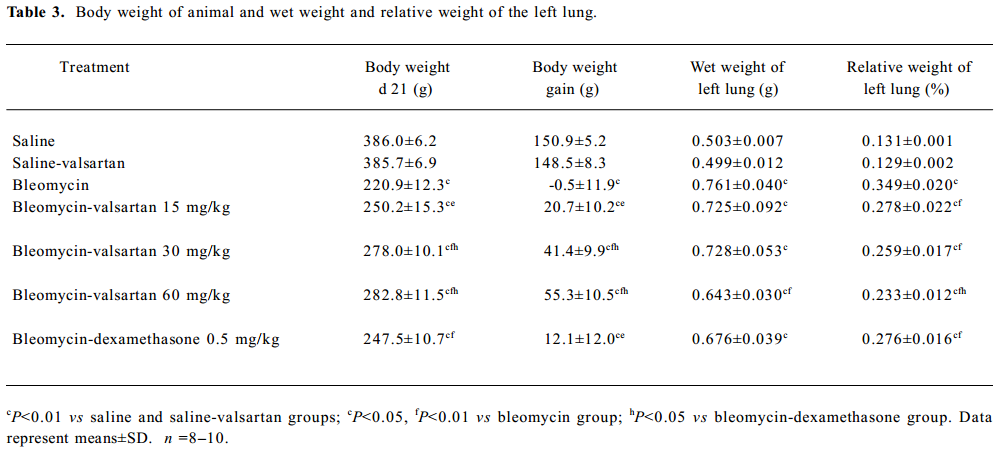
Effect of valsartan on mortality, body weight and lung weight in rats Table 2 shows that the mortality of rats in the bleomycin group was higher than that in the saline group, while that of the 3 valsartan groups and the dexamethasone group was lower than that of the bleomycin group. The mortality of the valsartan 30 and the 60 mg·kg-1·d-1 groups was 0. Table 3 presents the results of body weight. Rats only receiving bleomycin lost weight significantly after bleomycin injection, but the rats receiving dexamethasone (0.5 mg·kg-1·d-1) or valsartan (15, 30, 60 mg·kg-1·d-1) after being treated with bleomycin gained body weight. The body weight of the 3 bleomycin-valsartan-treated groups was more than that of bleomycin-dexamethasone-treated group on d 21, and the rats receiving 60 mg·kg-1·d-1 valsartan gained more body weight than the 2 other valsartan groups. The body weight of the control rats receiving valsartan and saline was similar (Table 3). The left lung was weighed and the relative weight (lung weight/body weight×100%) was calculated for each animal. The increase in wet weight of the left lung is one of the indices representing lung edema. As shown in Table 3, the wet and relative weights of the left lung increased significantly after bleomycin treatment. The increased relative weight of the left lung induced by bleo-mycin declined significantly following valsartan and dexamethasone treatment, and the decline of the rats treated with bleomycin and valsartan (30, 60 mg·kg-1·d-1) were obviously more than those with only bleomycin (P<0.05). These results indicated that this compound ameliorated lung edema, and this negative result caused by bleomycin and valsartan (60 mg·kg-1·d-1) was most apparent among the 3 doses of valsartan and dexamethasone.
Full table
Full table
Effect of valsartan on the degree of lung fibrosis Hydroxyproline content, an efficient index of collagen deposition, and collagen I/III mRNA expression were measured in the lung samples. As shown in Figure 1A, valsartan (60, 30, 15 mg·kg-1·d-1) and dexamethasone depressed the high content of hydroxyproline induced by bleomycin, and the inhibitory degree of valsartan showed a dose-dependent manner. On mRNA expression of collagen I/III, the high expression in the bleomycin group on d 21 was mitigated by 60 mg·kg-1·d-1 valsartan (Figure 1B).
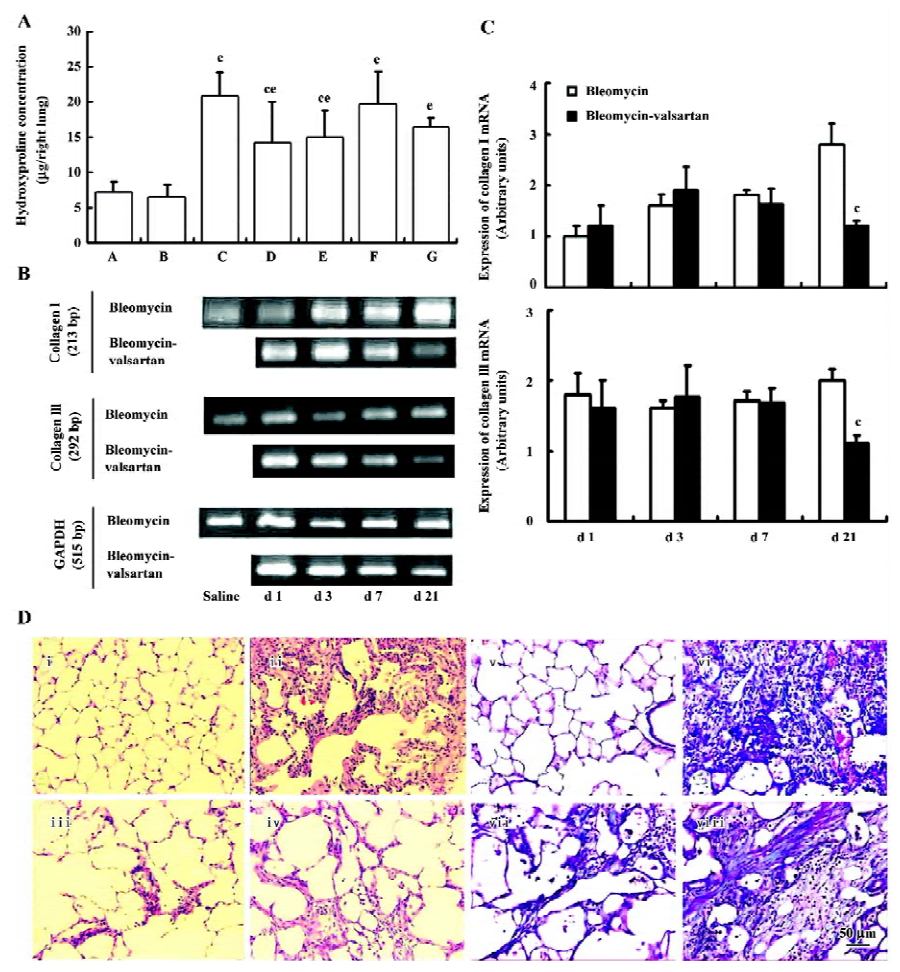
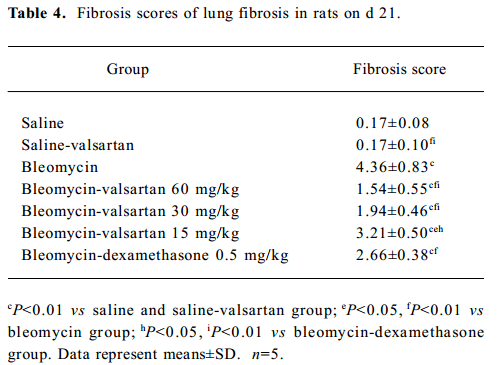
To further elucidate the histopathological changes associated with bleomycin-induced lung fibrosis and the efficacy of valsartan, the sections were stained with H&E and Masson trichrome. On d 21, marked histopathological changes, such as large fibrous areas, collapsed alveolar spaces, and traction bronchiectasis in the subpleural and peribronchial regions, were seen in the bleomycin-treated rats. Although fibrotic lesions were observed in the rats receiving bleomycin and valsartan (60 mg·kg-1·d-1), the extent of fibrosis was markedly less severe compared with that of the bleomycin group and bleomycin-dexamethasone group (Figure 1C). To confirm the effects of valsartan on the histopathology of bleomycin-induced lung fibrosis, the overall grades of fibrotic changes of the lungs were estimated by numerical scoring by blinding. As shown in Table 4, the 21-d treatment with 60 mg·kg-1·d-1 valsartan resulted in the depression of fibrosis induced by bleomycin significantly (P<0.01).
Full table
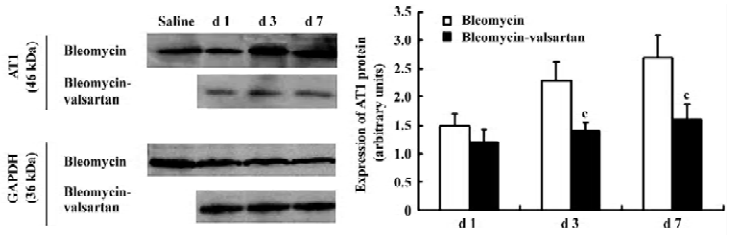
AT1 receptor in the early phase The level of AT1 receptor expression in the bleomycin group was measured in lung homogenates by Western blotting. In lung homogenates on d 1, 3, and 7, an anti-AT1 receptor antibody recognized a protein with an apparent molecular mass of 46 kDa. AT1 receptor expression located in lung tissues increased gradually from d 1 to 7 after bleomycin treatment (P<0.01; Figure 2).
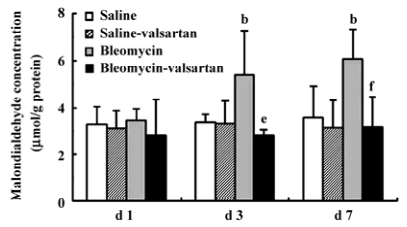
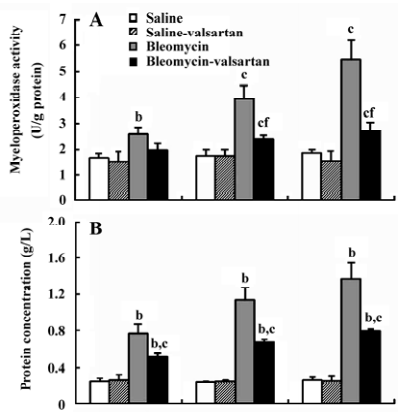
Lung injury in the early phase Malondialdehyde induction in the tissue samples and the increased protein concentration in BAL fluid are 2 other important markers of lung injury, with the first indicating the increased level of production of oxygen-free radicals and the latter indicating alveolar edema. Bleomycin-treated rats showed marked increases in malondialdehyde (Figure 3) and protein concentration (Figure 4); these levels peaked on d 7 after bleomycin treatment. Rats receiving valsartan (60 mg·kg-1·d-1) showed significantly lower increases in these levels. A similar situation was found with myeloperoxidase activity (Figure 4), alveolitis scores in lung tissue (Table 5), and the number of total inflammatory cells in BAL fluid (Table 6). The level of malondialdehyde concentration, protein concentration, myeloperoxidase activity and cell count were positively correlated with AT1 receptor expression in lung tissues in bleomycin-treated rats in the early stage, and valsartan reduced all of these increases. These data collectively indicate that the AT1 receptor contributes to lung injury induced by bleomycin.
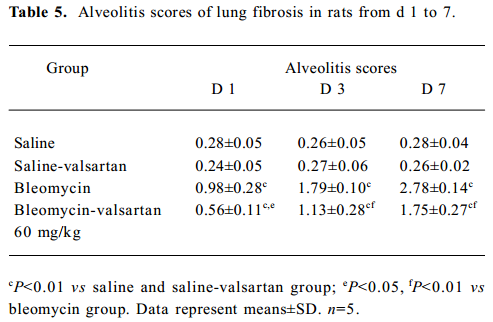
Full table
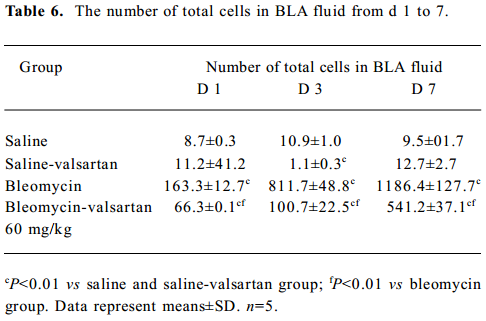
Full table
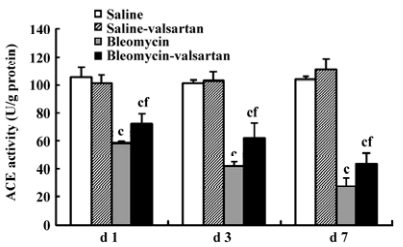
ACE activity in the early phase ACE activity is an index of pulmonary endothelial cell metabolic function, and decreased ACE activity has been proposed as an early indicator of bleomycin lung toxicity[22]. In the bleomycin group, the tendency of ACE activity declined gradually from d 1, and was lowest on d 7 (Figure 5). Valsartan reduced the decrease induced by bleomycin in the time course. These results implied that angiotensin II augmented bleomycin lung toxicity via the AT1 receptor.
Apoptosis in lung tissues in the early phase In the bleomycin-treated rats, the first event noted was endothelial damage of the lung vasculature, and apoptosis of AEC accompanied. Apoptosis in lung tissues from d 1 to d 7 was analyzed by in situ TUNEL assay. TUNEL-positive nuclei in the alveolar septum increased gradually from d 1 and were highest on d 7 in the bleomycin-treated rats (Figure 6A). There were obviously less apoptosis nuclei in the lung tissues of the valsartan-treated rats than the bleomycin-treated rats, especially on d 3 and d 7.
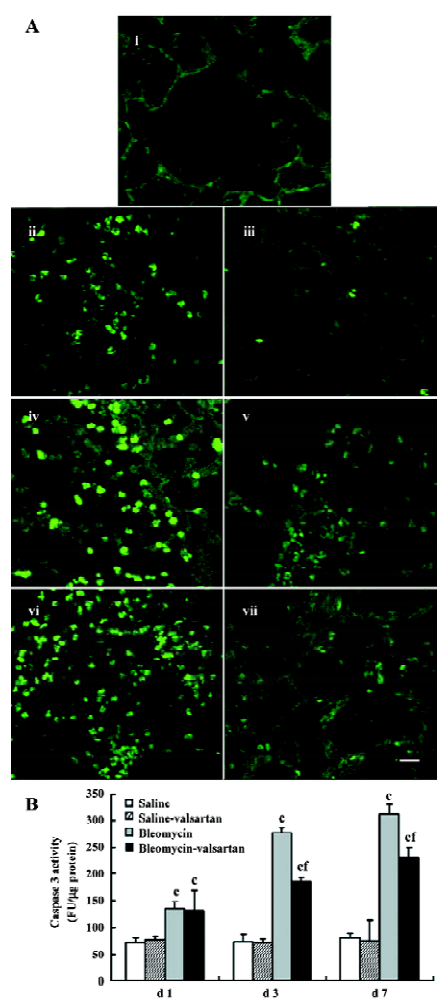
In addition, apoptosis was detected as an increase in the total activity of caspase-3 in the lung tissues, measured by enzyme assay of lung homogenates (Figure 6B). As early as the first day after instillation of bleomycin intratracheally, lung caspase-3 activity increased by 100%, and peaked on d 7, but the increase was prevented by the co-administration of valsartan.
Discussion
Inhibitors of ACE or antagonists of the AT1 receptor have been shown to have antifibrotic effects in the heart[23], kidney[24] and liver[25]. The first report of antifibrotic actions in the lungs by the ACE inhibitor captopril[26], published many years ago, was recently extended by the demonstration that the AT1 antagonists, losartan, L158809[16] and candesartan[9] have even more potent antifibrotic potential in the lung than ACE inhibitors. The present work extends those observations by showing that another AT1 antagonist, valsartan, also delays lung fibrosis induced by bleomycin in rats, and one of the mechanisms by which AT1 antagonists act is through the inhibition of lung injury in the early phase, including inflammation, apoptosis and oxidative stress.
Bleomycin-induced lung injury is associated with the recruitment of inflammatory cells into the injured lungs during the first 7 d[27]. This is accompanied by a rapid increase in pulmonary microvascular leak, which is manifested as an increase in total protein in BAL fluid[28]. Assessment of these early inflammatory responses in the present study was revealed by increases of total inflammatory cell number in BAL fluid in bleomycin-treated rats compared with saline-treated rats. Assessment of myeloperoxidase activity and alveolitis scores within the lungs revealed similar increases. Valsartan reduced the increases of total inflammatory cell number, alveolitis scores and myeloperoxidase activity, suggesting that the increase of the AT1 receptor may contribute to these increased responses in bleomycin-treated rats.
Earlier work has shown that AEC undergoing apoptosis in response to Fas ligand, tumor necrosis factor-α or bleomycin begin secreting angiotensin II into the extracellular space within hours of exposure, at least in vitro[29–31]. Those studies also implied that the autocrine production of angiotensin II and its binding to the AT1 receptor on AEC were required for apoptosis in response to these agents. In a previous study, the ACE inhibitor captopril or the caspase inhibitor ZVAD-fmk had essentially equal ability, blocking the appearance of apoptotic epithelial cells in rats exposed to intratracheal bleomycin and preventing subsequent collagen deposition, which suggests that the blockade of fibrogenesis by captopril is indeed related to the inhibition of apoptosis in vivo[11]. The present study, which shows that valsartan depressed the apoptosis from d 1 to d 7 and extracellular matrix in later phage induced by bleomycin, was consistent with the findings that valsartan is able to block both apoptosis and collagen deposition in rats. To capillary endothelial cells apoptosis, the action of angiotensin II were prevented in circulation and kidney via AT1 receptor inhibitors, although there is little paper studying in lung update, [32]. AT1 receptor signaling augments endothelial cells apoptosis in the process of oxidative stress-induced vascular injury[33]. The apoptosis of endothelial and epithelial cells in the lungs increases the permeability of the air-blood barrier and enhances the infiltration of inflammatory cells[34], which is one of mechanisms promoting lung fibrosis in idiopathic PF patients and bleomycin-treated rats[35,36]. ACE activity in located lung tissues, an index of pulmonary endothelial cell metabolic function[37], decreased in the early stage in the bleomycin-treated rats, which shows that endothelial cells in lung tissues were injured.
On the other hand, recent findings suggest that oxidative stress may play an important role in the pathogenesis of tissue fibrosis affecting apoptosis of both structural and inflammatory cells and altering the cytokine microenvironment balance[38,39]. It has been demonstrated that the presence of oxidative stress may lead to the damage, activation and/or apoptosis of AEC either directly, through an imbalanced intracellular redox equilibrium, or indirectly, by activating redox-sensitive effector pathways such as transcription factors and angiotensin-converting enzymes, increasing the conversion of angiotensinogen into angiotensin II which can be considered a mediator of oxidative stress[40]. These responses are correlated with high levels of malondialdehyde, which indicate that the increased lipid peroxidation is produced by oxygen-free radicals[41]. Data from this study revealed that bleomycin significantly increased malondial-dehyde production in lung tissues. This effect could be a secondary event, following a bleomycin-induced increase in free radical generation and/or decrease in lipid peroxidation protecting enzymes. Previous studies have reported that bleomycin-induced lung toxicity is related to redox cycling of an iron-bleomycin complex, which in turn catalyzes the formation of reactive oxidative species with ultimate progression of lipid peroxidation[42]. The results of this study show that valsartan attenuates increases in lung tissue malondialdehyde concentration induced by bleomycin, suggesting that angiotensin II is involved in oxidative stress in bleomycin-treated lung tissues via AT1.
The view that angiotensin II promotes human lung fibroblasts proliferation via the activation of the AT1 receptor in vitro was demonstrated by accumulating evidence[43]. A hypothesis testified that angiotensin II generation contributes directly to the fibroproliferative response to lung injury and collagen synthesis in vivo[44], but little was done with regards to the relationship of early lung injury and angiotensin II in bleomycin-induced lung fibrosis. In the present study, we found that in the early phase, bleomycin-induced apoptosis, inflammation and oxidative stress of lung tissue were significantly attenuated by the AT1 receptor antagonist, valsartan. These results show that angiotensin II also contributes to early lung injury, suggesting that a new mechanism of local renin-angiotensin system involved in the pathology of lung fibrosis. These studies are in agreement with the possibility that AT1 receptor antagonists may hold potential for the treatment of PF in humans.
References
- Kelley J. Cytokines of the lung. Am Rev Respir Dis 1990;141:765-88.
- Ward PA, Hunninghake GW. Lung inflammation and fibrosis. Am J Respir Crit Care Med 1998;157:S123-9.
- Geiser T. Idiopathic pulmonary fibrosis –– a disorder of alveolar wound repair? Swiss Med Wkly 2003;133:405-11.
- Cooper J Jr, White D, Matthay R. Drug-induced pulmonary disease: I. Cytotoxic drugs. Am Rev Respir Dis 1986;133:321-40.
- Thrall RS, Swendsen CL, Shannon TH, Kennedy CA, Frederick DS, Grunze MF, et al. Correlation of changes in pulmonary surfactant phospholipids with compliance in bleomycin-induced pulmonary fibrosis in the rat. Am Rev Respir Dis 1987;36:113-8.
- Thrall RS, McCormick JR, Jack RM, McReynolds RA, Ward PA. Bleomycin-induced pulmonary fibrosis in the rat: inhibition by indomethacin. Am J Pathol 1979;95:117-30.
- Murphy TJ, Takeuchi K, Alexander RW. Molecular cloning of AT1 angiotensin receptors. Am J Hypertens 1992;5:S236-42.
- Tanaka A, Matsumori A, Wang W, Sasayama S. An angiotensin II receptor antagonist reduces myocardial damage in an animal model of myocarditis. Circulation 1994;90:2051-5.
- Otsuka M, Takahashi H, Shiratori M, Chiba H, Abe S. Reduction of bleomycin induced lung fibrosis by candesartan cilexetil, an angiotensin II type 1 receptor antagonist. Thorax 2004;59:31-8.
- Bullock GR, Steyaert I, Bilbe G, Carey RM, Kips J, De Paepe B, et al. Distribution of type-1 and type-2 angiotensin receptors in the normal human lung and in lungs from patients with chronic obstructive pulmonary disease. Histochem Cell Biol 2001;115:117-24.
- Marshall RP. The pulmonary renin-angiotensin system. Curr Pharm Des 2003;9:715-22.
- Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT1 mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol 2002;282:L713-18.
- Specks U, Martin WJ II, Rohrbach MS. Bronchoalveolar lavage fluid angiotensin-converting enzyme in interstitial lung diseases. Am Rev Respir Dis 1990;141:117-23.
- Song L, Wang D, Cui X, Shi Z, Yang H. Kinetic alterations of angiotensin-II and nitric oxide in radiation pulmonary fibrosis. J Environ Pathol Toxicol Oncol 1998;17:141-50.
- Wang R, Alam G, Zagariya A, Gidea C, Pinillos H, Lalude O, et al. Apoptosis of lung epithelial cells in response to TNF-alpha requires angiotensin II generation de novo. J Cell Physiol 2000;185:253-9.
- Molteni A, Moulder JE, Cohen EF, Ward WF, Fish BL, Taylor JM, et al. Control of radiation-induced pneumopathy and lung fibrosis by angiotensin-converting enzyme inhibitors and an angiotensin II type 1 receptor blocker. Int J Radiat Biol 2000;76:523-32.
- Szapiel SV, Elson NA, Fulmer JD, Hunninghake GW, Crystal RG. Bleomycin-induced interstitial pulmonary disease in the nude, athymic mouse. Am Rev Respir Dis 1979;120:893-9.
- Brown S, Worsfold M, Sharp C. Microplate assay for the measurement of hydroxyproline in acid-hydrolyzed tissue samples. Biotechniques 2001;30:38-40, 42.
- Gong LK, Li XH, Wang H, Zhang L, Cai Y, Qi XM, et al. Feitai attenuates bleomycin-induced pulmonary fibrosis in rats. Biol Pharm Bull 2004;27:634-40.
- Unger T, Hubner D, Schull B, Yukimura T, Rascher W, Lang RE, et al. Dissociation between in vivo and in vitro measurements of converting enzyme activity after chronic oral treatment with captopril in rats. Am J Cardiol 1982;49:1530-2.
- Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem 1997; 272: 25 719–23.
- Takabatake N, Arao T, Sata M, Abe S, Inoue S, Shibata Y, et al. Involvement of pulmonary endothelial cell injury in the pathogenesis of pulmonary fibrosis: clinical assessment by 123I-MIBG lung scintigraphy. Eur J Nucl Med Mol Imaging 2005;32:221-8.
- Kokkonen JO, Saarinen J, Kovanen PT. Angiotensin II formation in the human heart: an ACE or non-ACE-mediated pathway? Ann Med 1998;30:9-13.
- Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension 2001;38:635-8.
- Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 2001;34:745-50.
- Molteni A, Ward WF, Ts’ao CH, Solliday NH, Dunne M. Monocrotaline-induced pulmonary fibrosis in rats: amelioration by captopril and penicillamine. Proc Soc Exp Biol Med 1985;180:112-20.
- Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol 2002;83:111-9.
- Hay J, Shahzeidi S, Laurent G. Mechanisms of bleomycin-induced lung damage. Arch Toxicol 1991;65:81-94.
- Wang R, Zagariya A, Ang E, Ibarra-Sunga O, Uhal BD. Fas induced apoptosis of alveolar epithelial cells requires ANG II generation and receptor interaction. Am J Physiol Lung Cell Mol Physiol 1999;277:L1245-50.
- Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal BD. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am J Physiol Lung Cell Mol Physiol 2000;279:L143-51.
- Li X, Zhang H, Soledad-Conrad V, Zhuang J, Uhal BD. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am J Physiol Lung Cell Mol Physiol 2003;284:L501-7.
- Lin LY, Lin CY, Su TC, Liau CS. Angiotensin II-induced apoptosis in human endothelial cells is inhibited by adiponectin through restoration of the association between endothelial nitric oxide synthase and heat shock protein 90. FEBS Lett 2004;574:106-10.
- Akishita M, Nagai K, Xi H, Yu W, Sudoh N, Watanabe T, et al. Renin-angiotensin system modulates oxidative stress-induced endothelial cell apoptosis in rats. Hypertension 2005;45:1188-93.
- Kuwano K, Hagimoto N, Nakanishi Y. The role of apoptosis in pulmonary fibrosis. Histol Histopathol 2004;19:867-81.
- Gerbitz A, Nickoloff BJ, Olkiewicz K, Willmarth NE, Hildebrandt G, Liu C, et al. A role for tumor necrosis factor-alpha-mediated endothelial apoptosis in the development of experimental idiopathic pneumonia syndrome. Transplantation 2004;78:494-502.
- Liu CY, Liu YH, Lin SM, Yu CT, Wang CH, Lin HC, et al. Apoptotic neutrophils undergoing secondary necrosis induce human lung epithelial cell detachment. J Biomed Sci 2003;10:746-56.
- Lazo JS, Lynch TJ, McCallister J. Bleomycin inhibition of angiotensin-converting enzyme activity from serum, lungs, and cultured pulmonary artery endothelial cells. Am Rev Respir Dis 1986;134:73-8.
- Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med 2005;172:417-22.
- Lenz AG, Hinze-Heyn H, Schneider A, Behr J, Haussinger K, Heindi S, et al. Influence of inflammatory mechanisms on the redox balance in interstitial lung diseases. Respir Med 2004;98:737-45.
- Mastruzzo C, Crimi N, Vancheri C. Role of oxidative stress in pulmonary fibrosis. Monaldi Arch Chest Dis 2002;57:173-6.
- Ullegaddi R, Powers HJ, Gariballa SE. Antioxidant supplementation enhances antioxidant capacity and mitigates oxidative damage following acute ischaemic stroke. Eur J Clin Nutr 2005;59:1367-73.
- Cross CE, Warren D, Gerriets JE, Wilson DW, Halliwell B, Last JA. Deferoxamine injection does not affect bleomycin-induced lung fibrosis in rats. J Lab Clin Med 1985;106:433-8.
- Marshall RP, McAnulty RJ, Laurent GJ. Angiotensin II is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am J Respir Crit Care Med 2000;161:1999-2004.
- Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol 2004;286:L156-64.