
EGCG inhibits cardiomyocyte apoptosis in pressure overload-induced cardiac hypertrophy and protects cardiomyocytes from oxidative stress in rats1
Introduction
Cardiac hypertrophy is a pathological response of the heart to chronic pressure or volume overload. Epidemiological studies revealed that cardiac hypertrophy was an independent risk factor for ischemic heart disease, arrhythmia, and sudden death. Moreover, the hypertrophic heart often leads to dilated cardiomyopathy, and eventually causes congestive heart failure after sustained overload. Therefore, it is important to determine the mechanism of the development of cardiac hypertrophy and prevent or treat it[1]. Recent evidence shows that cardiac apoptosis, although at a low level, is present in overload cardiac hypertrophy[2,3]. It was proposed that chronic, low level of cardiac myocyte apopto-sis is a causal component in the pathogenesis of heart failure. Cardiac apoptosis might be a critical factor during the transition from compensatory hypertrophy to heart failure[3,4]. The mechanisms of cardiac apoptosis include extrinsic factors and some cellular signal pathways. Bcl-2 expression has been proposed as an important marker of myocardial cell survival probability[5]. Tumor suppressor protein p53, a transcriptional modulator of the bcl-2 genes, can produce cell cycle arrest and facilitate apoptosis[5]. Condorelli et al[6] reported cardiomyocyte apoptosis contributed to the transition from pressure overload-induced cardiac hypertrophy to heart failure with dramatic downregulation of bcl-2, predisposing cardiomyocytes to apoptosis. Leri et al[7] reported the upregulation of local renin-angiotensin system in stretched cardiomyocytes increased susceptibility of myocytes to undergo apoptosis, coupled with the activation of p53 and bcl-2 decrease. In addition, in pressure overload-induced cardiac hypertrophy, excessive reactive oxygen species (ROS) may be generated in cardiac tissue. In vitro and in vivo studies have demonstrated that ROS may activate necrosis, apoptosis, even hypertrophy in cardiomyocytes[8,9].
Recently, more and more antioxidants have shown inhibitory effects on cardiac hypertrophy[10,11]. Epigallocate-chin gallate (EGCG), the major component of polyphenols in green tea, has recently attracted considerable attention for antioxidative, anti-inflammatory, antitumorigenic and antisenescent properties[12]. Recent research has shown that EGCG and green tea exerts protective effects against cardiovascular diseases[13–15]. Townsend et al[15] reported that green tea extracts and EGCG protected cardiomyocytes against ischemia/reperfusion-induced apoptotic cell death both in vivo and in vitro. More recently, results form both our laboratory[16] and from Li et al[17] showed that EGCG effectively inhibited cardiac hypertrophy in mice and rats. However, to date, little is known about the effects of EGCG on cardiac apoptosis in pressure overload-induced cardiac hypertrophy. It is known that EGCG may have an exceptional antioxidant capacity, even far exceeding than that of vitamin E and vitamin C[18]. We hypothesized that the inhibitory effects of EGCG on cardiac hypertrophy might be related to its antioxidant effects by scavenging ROS and inhibiting cardiac apoptosis in the hypertrophic myocardium.
To test this hypothesis, an abdominal aortic constriction model was established to detect the effects of EGCG on pressure overload-induced oxidative stress and cardiac apoptosis in cardiac hypertrophy with an analysis of p53 and bcl-2 protein expression in the hypertrophic myocardium. Mean-while, cultured newborn rat cardiomyocytes were exposed to exogenous hydrogen peroxide to determine the effects of EGCG on oxidative stress-induced cardiomyocyte injury and apoptosis in vitro.
Materials and methods
Materials EGCG used in animal experiments was purchased from Sichuan Leshan Yujia Tea Science and Technology Development Co Ltd (Leshan, Sichuan, China, purity >95%), while EGCG used in the cell experiments was purchased from Sigma (St Louis, USA). Captopril (Cap) was purchased from Jiangsu Huanghe Pharmaceutical Co Ltd (Jiangsu, China). Assay kits of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione peroxidases (GSH-Px) and lactate dehydrogenase (LDH) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The terminal transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay kit was purchased from Fujian Maixin Biotechnology Co Ltd (Fuzhou, Fujian, China). Mouse anti-p53 monoclonal antibody (sc-100) and rabbit anti-bcl-2 polyclonal (sc-492) antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA). Mouse anti-á-sarcomeric actin monoclonal antibody was purchased from Sigma (St Louis, USA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Carlsbad, California, USA).
Cardiac hypertrophy model induced by abdominal aortic constriction Male Sprague-Dawley rats (187.4±9.7 g) were obtained from the Experimental Animal Center of Soochow University (Suzhou, China; Certificate N
Determination of systolic blood pressure and heart weight indices Before the operation and each week after operation, systolic blood pressure (SBP) was measured between 10:00 and 12:00 AM in each rat by use of the tail cuff method after the rats were warmed at 37 °C for 30 min[17]. Six weeks after administration (7 weeks after AC), blood samples were collected from aorta for the detection of MDA, SOD, and GSH-Px; the rats were then sacrificed and the hearts were excised. After rinsing in phosphate buffered saline (PBS), the atria and right ventricular free wall were carefully dissected from the left ventricle. The wet weights of the whole heart and left ventricle were measured. The degree of cardiac hypertrophy was estimated by calculating the heart weight index (HWI£½HW/body weight (BW)) and left ventricular weight index (LVWI=LVW/BW)[19,20].
Detection of MDA content, and SOD and GSH-Px activities in serum and cardiac homogenates The blood samples were centrifuged at 1700×g for 10 min to obtain serum. The left ventricle was homogenized in 9 (v/v) volumes of ice-cold PBS, and the homogenates were centrifuged at 3000×g 4 °C for 15 min to obtain the supernate. MDA content, and SOD and GSH-Px activities in serum and cardiac homogenates were measured by assay kits, respectively, according to the manufacturer’s instructions[21].
Histological analysis The left ventricles were fixed in 10% formalin. The fixed hearts were embedded in paraffin, and sectioned at 4-µm thickness, then stained with hematoxylin and eosin or by van Gieson method.
TUNEL assay TUNEL reaction was performed using an in situ cell death detection kit, according to manufacturer’s instructions. Briefly, the section was deparaffinized and rehydrated with serial changes of xylene and ethanol. Proteinase K 20 mg/L was applied to the section for 15 min. The endogenous peroxidase was inhibited with 0.3% H2O2 for 5 min. The section was treated with the reaction mixture containing TdT and biotinylated 16-dUTP for 1 h at 37 °C. Labeled DNA was visualized with peroxidase-conjugated antidigoxigenin antibody using 3,3'-diaminobenzidine as the chromogen. TUNEL positive cells were detected according to the instructions. Ten random fields per section were analyzed, respectively, and the average value of the TUNEL positive cardiomyocyte percentage was calculated[22,23].
Western blot analysis The total cardiac protein was extracted and Western blot analysis was performed as described previously[6,24]. Briefly, the left ventricular tissues were homogenized in a buffer containing Tris-HCl (pH 7.4) 10 mmol/L, NaCl 150 mmol/L, 1% Triton X-100, 1% sodium deoxycho-late, 0.1% SDS, edetic acid 5 mmol/L, phenyl-methylsulfonyl fluoride (PMSF) 1 mmol/L, aprotinin 0.28 kU/L, leupeptin 50 mg/L, benzamidine 1 mmol/L, and pepstatin A 7 mg/L. Protein concentration was determined by a bicin-choninic acid (BCA) kit (Pierce, Rockford, IL, USA). Eighty milligrams of protein from each sample was loaded onto 12% SDS-PAGE gel and subjected to electrophoresis using a constant current. The proteins were transferred to the nitrocellulose membranes (Amersham, Arlington Height, IL, USA) and incubated with rabbit anti-bcl-2 polyclonal antibody or mouse anti-p53 monoclonal antibody in Tris buffered saline containing 0.1% Tween-20 (TBST) and 5% nonfat dry milk for 3 h. The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz, USA) in TBST containing 5% nonfat dry milk for 1 h. Immunoreactivity was detected by enhanced chemolumine-scent autoradiography (ECL kit, Amersham, USA) according to manufacturer’s instructions. The results were analyzed quantitatively using SigmaScan Pro 5.0.0 (SPSS Inc, Chicago, Illinois, USA). The data were normalized with respect to the ratios of actin detected on the same blot to control for possible variations in protein loading.
Cell culture Newborn Sprague-Dawley rats, 1–2 d old, were put into PBS after disinfecting. The heart tissues were removed and put into cold D-Hanks’ solution. After the removal of the blood vessels and atria, the ventricles were minced and incubated in 0.25% trypsin at 37 °C for 10 min. The supernate was discarded and the tissue was redigested with trypsin for 10 min; an ice bath was then used to terminate digestion, and the supernate was centrifuged at 700×g for 10 min. The sediment was resuspended in DMEM containing 10% fetal bovine serum. The residual tissues were redigested for 4–6 cycles until single cell was obtained. Cell suspensions were collected together and preplated in culture flasks for 45 min at 37 °C in 95% air/5% CO2 to remove fibroblasts. The unattached cells were counted and seeded onto another culture flask at a density of 1.5×106/L. The medium was changed every 48 h. All experiments were performed on 3–5 d cultures when synchronously contracting cells were observed. The purity of the cardiomyocytes was confirmed by anti-α sarcomeric actin antibody[25,26].
Cell viability analysis The cardiomyocytes cultured in 96-well plates were preincubated with EGCG 12.5–200 mg/L for 6, 12, 24, and 48 h. H2O2 200 µmol/L was added and incubated with the cells for additional 24 h. After separating with the medium, the cells were washed twice with PBS. DMEM containing MTT 0.5 g/L was appended to each well and incubated at 37 °C for an additional 4 h. After the medium was discarded, the remaining formazan crystals were dissolved in 100 µL Me2SO. Absorbance at 570 nm was measured by a DG3002 ELISA plate reader (Huadong Electronic Company, Nanjing, China).
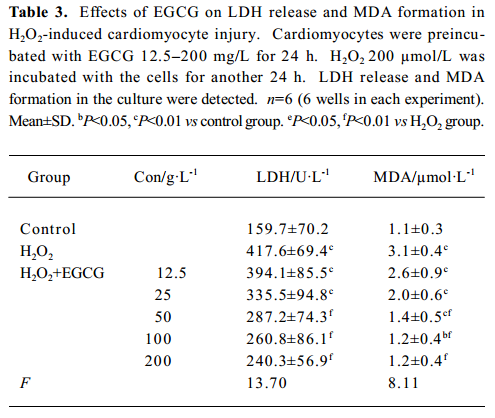
LDH release and MDA formation The cardiomyocytes cultured in 24-well plates were preincubated with EGCG 12.5–200 mg/L for 24 h; H2O2 200 µmol/L was added and incubated with the cells for another 24 h. LDH activity in the medium was measured by a LDH assay kit at 440 nm. MDA content in the medium was measured by a MDA assay kit at 532 nm[27].
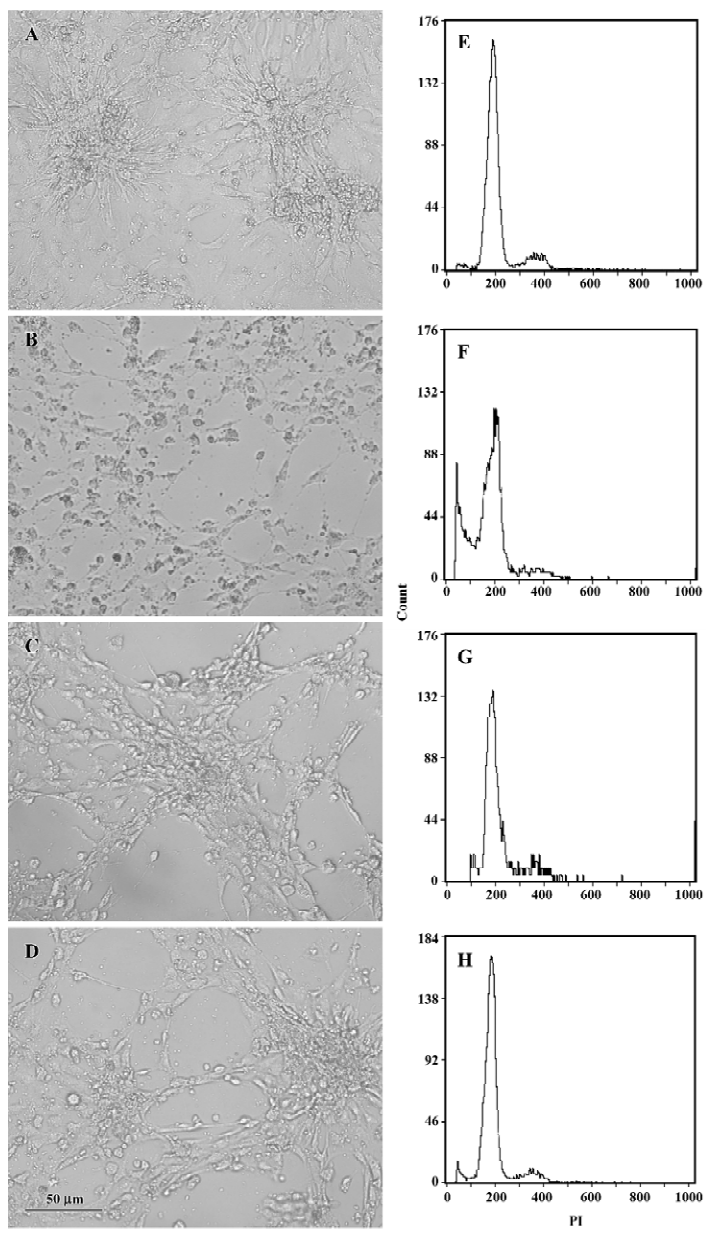
Flow cytometry analysis After pretreatment with EGCG 50 and 100 mg/L for 24 h, H2O2 200 µmol/L was added and incubated with the cells for an additional 24 h. Cell morphological changes were observed by a phase contrast micro-scope. For the flow cytometry analysis, the cells (>1×106) were digested with 0.25% trypsin and collected by centri-fugation. After washing twice with ice-cold PBS, the cells were fixed in ice-cold 70% ethanol. After centrifugation, the fixed cells were incubated with RNase 100 mg/L at 37 °C for 30 min and stained with 50 mg/L propidium iodide (PI) for 30 min. The cells were analyzed by EPICS XL flow cytometry (Beckman Coutler, Califonia, USA). The hypodiploid population of cells was considered apoptosis, and the apoptotic rate was analyzed by Multicycle software (Beckman Coutler, Califonia, USA)[28].
Statistical analysis Data were expressed as mean±SD. One-way ANOVA in SPSS 10.0 software (SPSS Inc, Chicago, Illinois, USA) was used for the statistical analysis. The intergroup comparisons (post-hoc analysis) among the data with equal variances were made by the LSD method, while Tamhane’s T2 method was used for the data with unequal variances.
Results
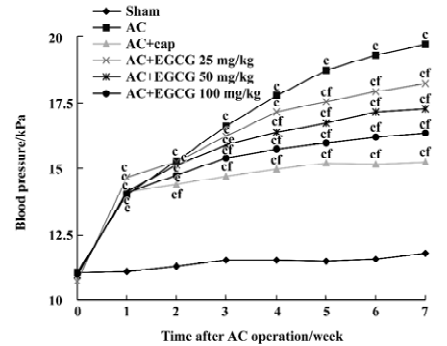
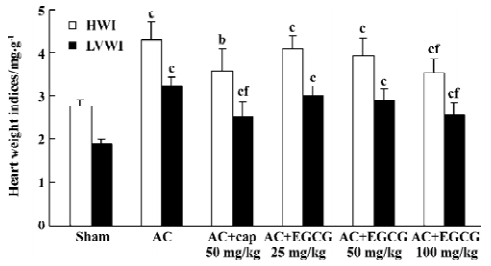
Effects of EGCG on SBP and heart weight indices In the rats with cardiac hypertrophy, compared with the sham-operated group, the SBP of AC rats began to increase 1 week after the operation, and increased progressively at 2–7 weeks after the operation (Figure 1). The heart weight indices also increased remarkably vs the sham-operated group. The HWI and LVWI increased by 55.8% and 72.3%, respectively (Figure 2). However, treatment with EGCG 25, 50, and 100 mg/kg for 6 weeks reduced SBP in a time-dependent manner (F=0.2, 14.3, 52.9, 67.1, 64.3, 80.0, 100.0, 111.2 from 0 to 7 weeks postoperation, respectively). EGCG chronic treatment also reduced HWI and LVWI compared with the AC group (F=21.0 and 35.1, respectively). The HWI was reduced by 4.7%, 8.37%, and 17.7% at 3 doses respectively, whereas the LVWI was reduced by 7.1%, 11.1%, and 21.3%, respectively.
Effects of EGCG on MDA content, and SOD and GSH-Px activities in serum and the myocardium In the rats with cardiac hypertrophy, the level of MDA content was increased, and SOD and GSH-Px activities were reduced significantly vs the sham-operated group, both in serum and in the myocardium (P<0.01; Tables 1, 2). EGCG 25, 50, and 100 mg/kg dose-dependently decreased MDA content, and increased SOD and GSH-Px activities in serum and the myocardium compared with the AC group.
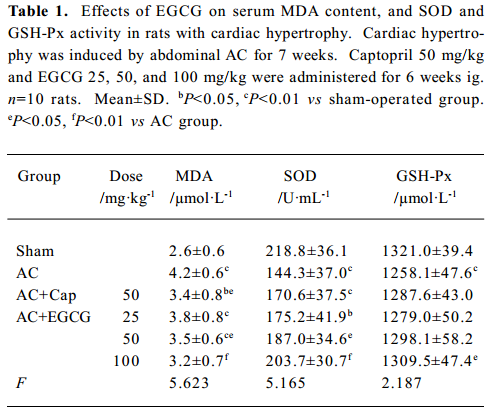
Full table
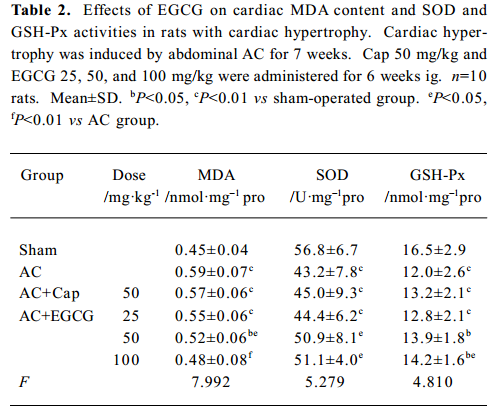
Full table
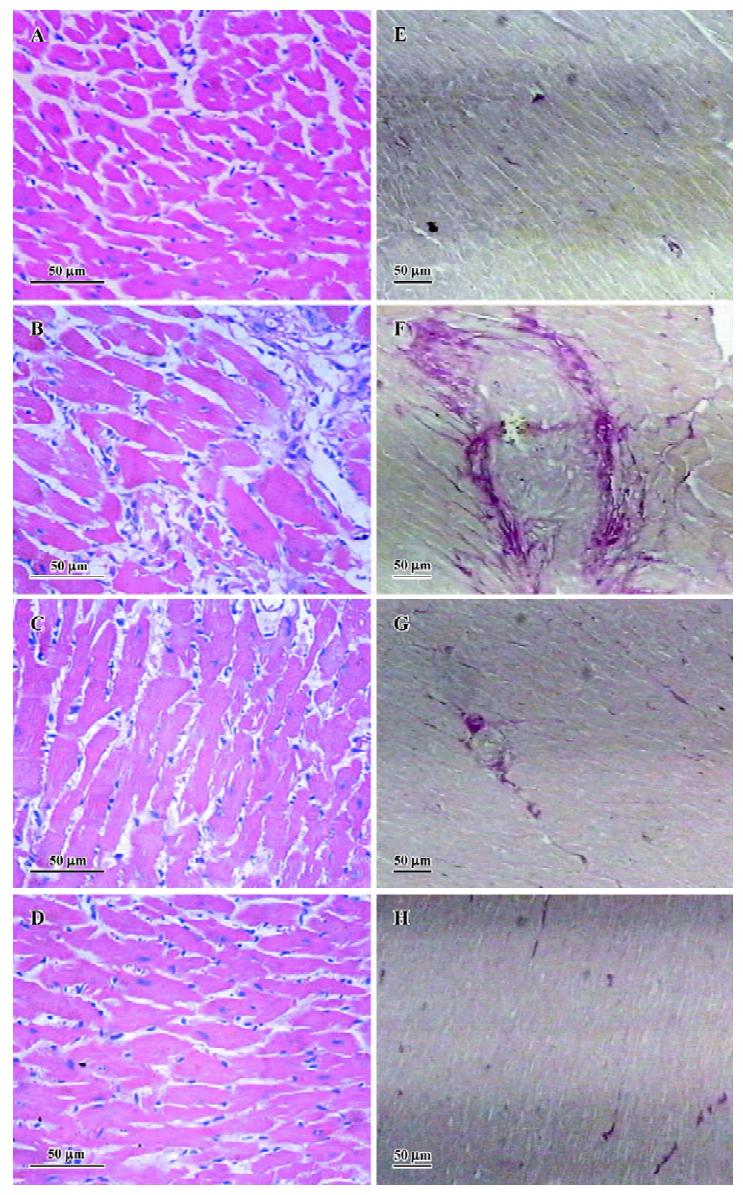
Effects of EGCG on histological changes HE stain of the heart tissues showed that compared with sham-operated group, the hearts of the AC group displayed marked structural abnormalities, cardiomyocyte hypertrophy, and cellular fibrosis (Figure 3A–3D). Treatment with EGCG 50 and 100 mg/kg remarkably improved the histological changes in the heart tissue vs the AC group. As demonstrated by VG stain, significant intermuscular fibrosis was observed in the hypertrophic hearts, while EGCG 50 and 100 mg/kg treatment diminished the extent of fibrosis (Figure 3E–3H).
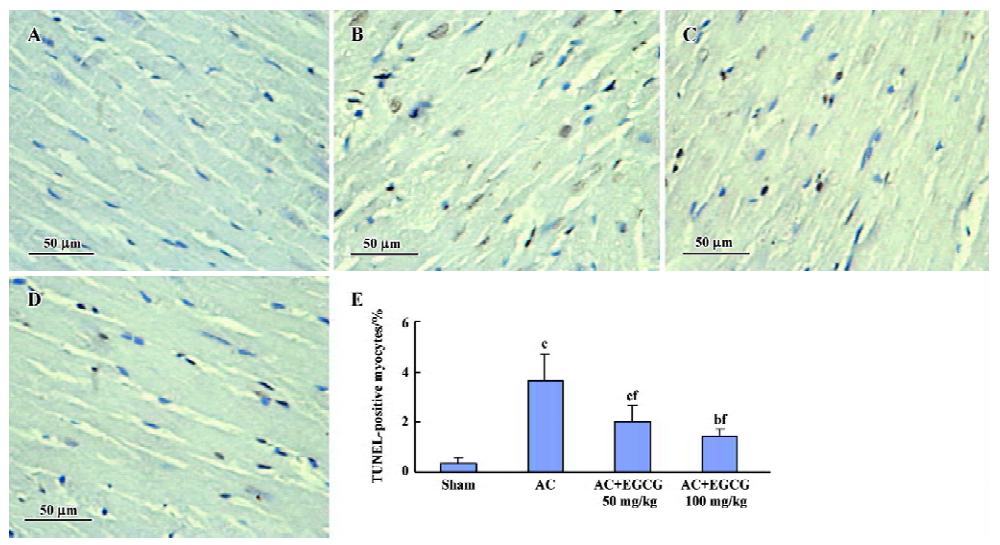
Effects of EGCG on cardiac apoptosis in the hypertrophic myocardium In the hearts of the sham-operated group, TUNEL-positive cells were nearly undetectable, but numerical TUNEL-positive cells were observed in the myocardium of rats with cardiac hypertrophy. Treatment with EGCG 50 and 100 mg/kg for 6 weeks showed a significant reduction in the number of TUNEL-positive cells compared with the AC rats (F=21.2; Figure 4).
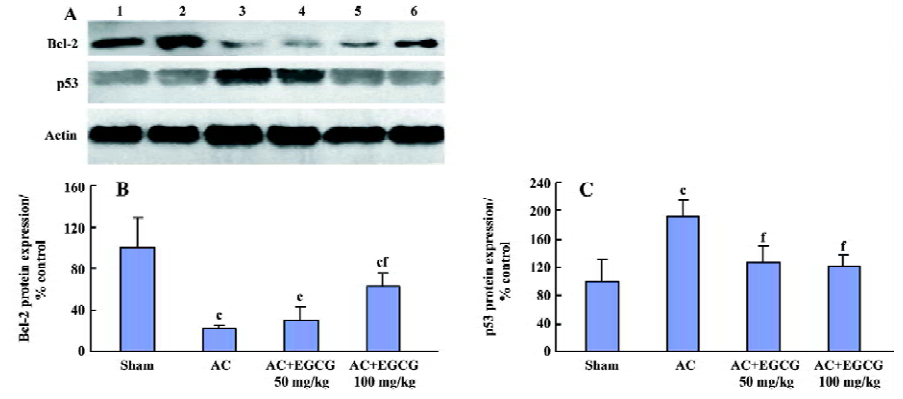
Effects of EGCG on p53 and bcl-2 protein expression in the myocardium In the rats with cardiac hypertrophy, the level of the p53 protein increased, and the bcl-2 protein decreased remarkably compared with the sham-operated group. EGCG 50 and 100 mg/kg ig for 6 weeks decreased p53 protein expression and increased bcl-2 protein expression in the hypertrophic myocardium vs the model group (F=30.8 and 29.2, respectively; Figure 5).
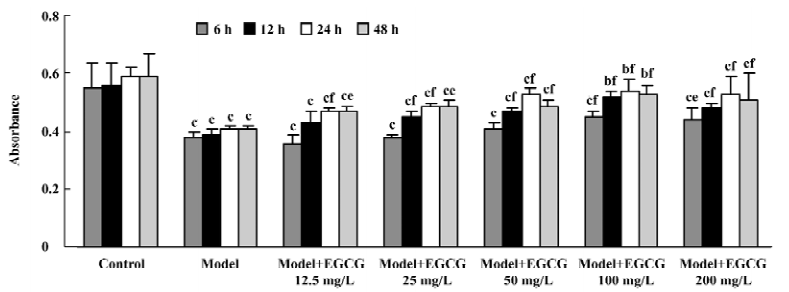
Effects of EGCG on cell viability in H2O2-induced injury The exposure of cardiomyocytes to H2O2 200 µmol/L for 24 h produced an obvious decrease in cell viability as measured by MTT (P<0.01 vs the model group). When the cultures were pretreated with EGCG 12.5–200 mg/L for 6–48 h, the cell damage was greatly attenuated (F=13.9, 13.5, 19.3, 7.5 from 6–48 h, respectively; Figure 6). The protective effects of EGCG were shown at pretreatment of myocytes for 6 h, reached the best at 24 h, and maintained its effect at 48 h. As to the concentration, pretreatment with EGCG 12.5–100 mg/L concentration-dependently inhibited H2O2-induced cardiomyocyte injury, but the effect of EGCG 200 mg/L was reduced to a degree.
Effects of EGCG on LDH release and MDA formation The exposure of cardiomyocytes to H2O2 200 µmol/L for 24 h induced marked MDA formation and LDH leakage in the culture medium (P<0.01; Table 3). Pretreatment with EGCG 12.5–200 mg/L for 24 h attenuated LDH release and MDA formation in the culture medium.
Full table
Effects of EGCG on H2O2-induced cardiomyocyte apoptosis When exposed to H2O2 200 µmol/L for 24 h, the sizes of most cardiomyocytes were reduced obviously. Cytoplasm shrinkage and nuclei pyknosis were found under microscope, indicating distinct apoptosis of cardiomyocytes. Pretreatment with EGCG 50 and 100 mg/L significantly attenuated the morphological changes in cardiomyocytes (Figure 7A–7D). When cardiomyocyte apoptosis was quantified by flow cytometry, the percentage of apoptotic cells increased from 0.8%±0.7% in the control group to 15.9%±2.3% in the H2O2 group (n=3, P<0.01 vs the control group; Figure 7E−7F). Pretreatment with EGCG 50 and 100 mg/L for 24 h decreased the apoptotic rate to 6.3%±3.1% and 3.6%±0.7%, respectively (F=32.08, P<0.01 vs the H2O2 group; Figure 7G–7H).
Discussion
Tea is one of the most consumed beverages in the world, especially in Asian countries. Tea consumption may be linked to low incidences of various pathological conditions, including cardiovascular disease, diabetes, obesity, and cancer. The principal active polyphenols in green tea include EGCG, epigallocatechin (EGC), epicatechin (EC) and epicatechin gallate (ECG), with EGCG being the most abundant, and possessing the most potent antioxidative activity. In our experiment, EGCG 25–100 mg/kg, administered once a day for 6 weeks was used to treat rats with cardiac hypertrophy. The dose was equivalent to the EGCG content in approximately 8−23 cups (one cup=120 mL) of green tea consumption everyday in humans (approximately 80 kg weight)[29].
Chronic pressure overload is the critical factor leading to cardiac hypertrophy and even heart failure. In the abdominal aortic constriction model, aortic pressure increased immediately after AC, and cardiac hypertrophy occurred as an adaptive response to the imposition of long-term pressure overload on the heart[19,20,30]. In the present study, the cardiac hypertrophy model was established by AC in the rats for 7 weeks. The SBP increased progressively at 1–7 weeks after aortic abdominal constriction. The HWI and LVWI increased remarkably vs the sham-operated group. Structural abnormality, cardiomyocyte hypertrophy, and fibrosis were detected in the hypertrophic myocardium by histological analysis. However, chronic treatment with EGCG 25–100 mg/kg dose-dependently reduced SBP, HWI, and LVWI, and improved histological changes in the myocardium. All these data supported our finding that EGCG was an effective therapeutic agent against pressure overload-induced cardiac hypertrophy.
In this paper, the effects of EGCG on rats with cardiac hypertrophy were compared with the AC group, so we did not detect the effect of EGCG on sham-operated rats. It is a limitation of the experiment and we will try to investigate it in the further experiments. Nevertheless, Li et al reported[17] the beneficial effects of EGCF on hemodynamics in the AC rat model with little effect on the sham-operated rats.
Mounting evidence strongly implicated that oxidative stress played an important role in the genesis and process of cardiac hypertrophy[10,11]. Oxidative stress leads to the accumulation of lipid peroxidation products MDA in the heart, and causes impaired cell function, while antioxidant enzyme SOD and GSH-Px play great roles in cellular defense against oxidative stress. In this study, to confirm the presence of increased oxidative stress in cardiac hypertrophy, we quantified myocardial and serum levels of MDA content, and SOD and GSH-Px activities. The results showed that in the pressure overload-induced cardiac hypertrophy rat model, the level of MDA in serum and cardiac tissue increased significantly, while the activities of SOD and GSH-Px was greatly reduced vs the sham-operated control, indicating a significant oxidative stress in cardiac hypertrophy. The treatment with EGCG almost completely prevented the pressure overload-induced decrement in SOD and GSH-Px levels, and MDA formation both in serum and in cardiac tissue. These results suggest that the protective effects of EGCG on cardiac hypertrophy were correlated with amelioration of pressure overload-induced oxidative stress.
As already stated, apoptosis is a critical factor during transition from compensatory cardiac hypertrophy to heart failure[4–6]. The introduction of the TUNEL method to localize the 3' end of DNA in situ has been proposed as a useful tool for the identification of apoptosis. It has been reported that pressure overload in the murine heart leads to an increase in TUNEL-positive cardiomyocytes[2,6]. In the present study, a TUNEL analysis was used to detect apoptotic myocytes in the hypertrophic myocardium. The results showed that in the pressure overload-induced cardiac hypertrophy rat model, TUNEL- positive cells increased significantly compared with the sham-operated group, while EGCG 50 and 100 mg/kg reduced the number of TUNEL-positive cells remarkably. We also tested whether EGCG could protect against cultured cardiomyocyte injury and apoptosis from oxidative stress. H2O2, one major kind of ROS, can lead to the formation of hydroxyl radicals mediated by intracellular heavy metal ions through the Fenton reaction. Both H2O2 and hydroxyl radicals induce severe intracellular oxidant stress, which cause damage to various intracellular biomacro-molecules and eventually result in apoptosis and necrosis of cardiomyocyte[31]. In this experiment, when cardiomyocytes were exposed to exogenous H2O2, cell viability decreased significantly, and marked LDH release was observed, indicating injury in the membrane integrality. Lipid peroxidation products MDA formation greatly increased, reflecting a significant oxidative stress. Pretreatment with EGCG 12.5–200 mg/L greatly increased cell viability, reduced LDH release and MDA formation in the culture medium. These results suggest that EGCG exerted its antioxidant effects in H2O2-induced cardiomyocyte injury. Moreover, H2O2 induced distinct apoptosis in cardiomyocytes. Morphological observation found cytoplasm shrinkage and nuclei pyknosis, and the apoptotic rate increased markedly, while preincubation with EGCG 50 and 100 mg/L significantly improved myocyte morphological changes and reduced the apoptotic rate in cardiomyocytes, indicating that EGCG attenuated cardiomyocyte apoptosis induced by oxidative stress.
Townsend et al[15] reported that green tea extract, as well as EGCG, prevented ischemia/ reperfusion (I/R)-induced cardiac myocyte apoptotic cell death, partly by reducing the expression of the STAT-1 proapoptotic target gene and the Fas receptor. However, in this paper we aimed at p53 cell death signal in cardiac hypertrophy rather than the death receptor pathway. Evidence shows that p53 plays an important role in cardiac myocyte apoptosis in cardiac hyper-trophy. Leri A et al[32] reported that the activation of the p53 and p53-dependent genes were critical in the modulation of myocyte apoptosis in pacing-induced heart failure. The upregulation of the local renin-angiotensin system in stretched cardiomyocytes increased susceptibility of myocytes to undergo apoptosis, coupled with the activation of p53 and bcl-2 decrease[7]. To determine whether stretch-induced activation of p53 is necessary for the upregulation of the local renin-angiotensin system and angiotensin (Ang) II-induced apoptosis, they infected myocytes with Adp53m to prevent p53 stimulation, and found Adp53m interference blocking the functions of endogenous wild-type p53, reducing Ang II generation and myocyte apoptosis after mechanical stretching. The research presents definitive proof that p53 is an essential cofactor in the stimulation of proapoptotic genes and the induction of cell death by mechanical stress[33]. Therefore, we used Western blot analysis to detect p53 and bcl-2 protein expression in the hypertrophic myocardium. Results showed a significant decrease in bcl-2 and an increase in the p53 protein in the hypertrophic myocardium compared with the sham-operated control. In contrast, EGCG treatment effectively upregulated the bcl-2 protein and downregulated the p53 protein. It has been reported that p53 can act to regulate the intracellular redox state and induce apoptosis by a pathway dependent on ROS production[5,8,34], whereas bcl-2 may function as an antioxidant to prevent apoptosis[5]. Bcl-2 may decrease lipid peroxidation by increasing cell resistance to ROS and blocking ROS production[5]. Therefore, we proposed the antiapoptotic effect of EGCG by inhibiting p53 induction and bcl-2 decrease might be related, at least in part, to its antioxidant effects. We tried to use RNA silencing technology to detect the effects of EGCG on p53-mediated apoptosis, but failed to silence the gene because of the difficulty of transfection into primary neonatal rat cardiomyocytes. Further investigation is needed to verify this proposal.
In summary, EGCG remarkably inhibits the formation of cardiac hypertrophy with marked reductions in pressure overload-induced oxidative stress and cardiac apoptosis. Also, EGCG protects cultured cardiomyocytes from hydrogen peroxide-induced injury and apoptosis. The mechanism might be related to the inhibitory effects of EGCG on p53 induction and bcl-2 decrease.
References
- Olivetti G, Cigola E, Maestri R, Lagrasta C, Corradi D, Quaini F. Recent advances in cardiac hypertrophy. Cardiovasc Res 2000;45:63-7.
- Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, et al. Apoptosis in pressure overload induced heart hypertrophy in the rat. J Clin Invest 1996;97:2891-7.
- Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol 1997;272:H2313-9.
- Wencker D, Chandra M, Nguyen K, Miao WF, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 2003;111:1497-504.
- Li PF, Dietz R, Harsdorf RV. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by bcl-2. EMBO J 1999;21:6027-36.
- Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G. Increased cardiomyocyte apoptosis and changes in proapo-ptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 1999;99:3071-8.
- Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the bcl-2-to-bax protein ratio in the cell. J Clin Invest 1998;101:1326-42.
- Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 2001;89:279-86.
- Chen QM, Tu VC, Wu Y, Bahl JJ. Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyo-cytes. Anch Biochem Biophys 2000;373:242-8.
- Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 1998;98:794-9.
- Tsujimoto I, Hikoso S, Yamaguchi O, Kashiwase K, Nakai A, Takeda T, et al. The antioxidant edaravone attenuates pressure overload-induced left ventricular hypertrophy. Hypertension 2005;45:921-6.
- Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Crit Rev Food Sci Nutr 2003;43:89-143.
- Chyu KY, Babbidge SM, Zhao X, Dandillaya R, Rietveld AG, Yano J, et al. Differential effects of green tea-derived catechin on developing versus established atherosclerosis in apolipoprotein E-null mice. Circulation 2004;109:2448-53.
- Priyadarshi S, Valentine B, Han C, Fedorova OV, Bagrov AY, Liu J, et al. Effect of green tea extract on cardiac hypertrophy following 5/6 nephrectomy in the rat. Kidney Int 2003;63:1785-90.
- Townsend PA, Scarabelli TM, Pasini E, Gitti G, Menegazzi M., Suzuki H, et al. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J 2004;18:1621-3.
- Sheng R, Gu ZL, Xie ML, Guo CY, Zhou WX. EGCG inhibits collagen accumulation and cell proliferation in cardiac hyper-trophy. Chin Pharmacol Bull 2006;22:1095-9.
- Li HL, Huang Y, Zhang CN, Liu G, Wei YS, Wang AB, et al. Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and -independent signal pathways. Free Rad Biol Med 2006;40:1756-75.
- Rice-Evans C. Implications of the mechanisms of action of tea polyphenols as antioxidants in vitro for chemoprevention in humans. Proc Soc Exp Biol Med 1999;220:262-6.
- Shimoyama M, Hayashi D, Takimoto E, Zou YZ, Oka T, Uozumi H, et al. Calcineurin plays a critical role in pressure overload–induced cardiac hypertrophy. Circulation 1999;100:2449-54.
- Li JL, Li P, Feng XH, Li ZP, Hou R, Han C, et al. Effects of lorsartan on pressure overload-induced cardiac gene expression profiling in rats. Clin Exp Pharmacol Physiol 2003;30:827-32.
- Yamamoto M, Yang GP, Hong C, Liu J, Holle E, Yu XZ, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest 2003;112:1395-406.
- Baldi A, Abbate A, Bussani R, Melfi GR, Angelini A, Dobrina A, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol 2002;34:165-74.
- Ji ES, Yue H, Wu YM, He RR. Effects of phytoestrogen genistein on myocardial ischemia/reperfusion injury and apoptosis in rabbits. Acta Pharmacol Sin 2004;25:306-12.
- Cao Y, Gu ZL, Lin F, Han R, Qin ZH. Caspase-1 inhibitor Ac-YVAD-CHO attenuates quinolinic acid-induced increases in p53 and apoptosis in rat striatum. Acta Pharmacol Sin 2005;26:150-4.
- Aikawa R, Nawano M, Gu YP, Katagiri H, Asano T, Zhu WD, et al. Insulin prevents cardiomyocytes from oxidative stress–induced apoptosis through activation of pI3 Kinase/Akt. Circulation 2000;102:2873-9.
- Akao M, Ohler A, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ Res 2001;88:1267-75.
- Zhang RL, Pinson A, Samuni A. Both hydroxylamine and nitroxide protect cardiomyocytes from oxidative stress. Free Rad Biol Med 1998;24:66-75.
- Sawyer DB, Fukazawa R, Arstall MA, Kelly RA. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazo-xane. Circ Res 1999;84:257-65.
- Chow SH-H, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Caner Res 2003;9:3312-9.
- Hong Y, Hui SSC, Chan BTY, Hou JY. Effect of berberine on catecholamine levels in rats with experimental cardiac hyper-trophy Life Sci 2003;72:2499-507.
- Harsdorf RV, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 1999;99:2934-41.
- Leri A, Liu Y, Malhotra A, Li Q, Stiegler P, Claudio PP, et al. Paing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation 1998;97:194-203.
- Leri A, Fiordaliso F, Setoguchi M, Limana F, Bishopric NH, Kajstura J, et al. Inhibition of p53 function prevents rennin-angiotensin system activation and stretch-mediated myocyte apoptosis. Am J Pathol 2000;157:843-57.
- Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA 1996;93:11848-52.