
One-oligonucleotide method for constructing vectors for RNA inter-ference1
Introduction
RNA interference (RNAi) is a powerful tool for inhibiting gene expression in a wide variety of organisms. When double-stranded RNA is introduced into cells, the ribonuclease III Dicer processes the double-stranded RNA into small fragments of about 21 nucleotides in length (termed siRNAs) that trigger the RNAi mechanism[1–3]. Then, the siRNAs are incorporated into a protein complex, known as the RNA-induced silencing complex (RISC), which in turn unwinds the duplex siRNA in an ATP-dependent manner[4]. After the unwinding, RISC uses the siRNA antisense strand as a guide to specifically cleave the complementary mRNA, which is further processed for degradation[4–6].
There are different ways to induce mRNA degradation using RNAi. Several methods for preparing siRNA have been developed, such as chemical synthesis, in vitro transcription, siRNA expression vectors, and polymerase chain reaction (PCR) expression cassettes. For example, short hairpin RNAs (shRNAs) transcribed by RNA polymerase III promoter-based vectors have been used as RNAi triggers in a variety of cell lines[7,8]. These RNA polymerase III (RNA Pol) vectors have a variety of advantages over the siRNAs. First, they greatly reduce the cost of RNAi synthesis, making RNAi a viable tool for screening the function of large numbers of genes. Second, because hairpins are transcribed inside cells, it is possible to establish inducible systems for RNAi both in vitro and in vivo[9–11]. Third, it is also possible to generate knock-down phenotypes to resemble knock-out animals without affecting the germline, as long as this strategy is combined with inducible systems[12–14]. Therefore, it is relatively easy to establish cell cultures using retroviral or adenoviral vectors that stably express the hairpin for RNAi[10,15–18].
Although there has been much progress in the RNAi field, it is not completely understood why some sequences are refractory to RNAi. Some groups have investigated this lack of activity, but at present, choosing the most effective siRNA sequence to knock down an mRNA is still a trial-and-error task. Thus, it is generally accepted that several sequences must be designed to achieve the highest RNAi efficiency, making the silencing experiments costly and slow. It would be advantageous to have a more efficient method for the production of a wide variety of sequences in a simple step, instead of using long DNA oligonucleotides or several PCR step-specific methods[8,19–21].
Here we describe an inexpensive, easy to implement and automated method to produce DNA cassettes for RNAi experiments using a single oligonucleotide. With this method hundreds of DNA cassettes can be synthesized in a 1-step reaction. This method not only reduces the cost of testing sequences for RNAi experiments, but also accelerates the exploration of multiple-gene sequences.
Materials and methods
Construction of DNA cassettes and plasmid vectors To construct the plasmid vector pBB4H1, the human H1 promoter was PCR amplified from human genomic DNA using the following primers: forward 5'-CCATGGAATTCGAA-CGCTGACGT-3' and reverse 5'-GC
To generate the DNA cassettes the following chemically synthesized oligonucleotides were used: hpGFP1 5'-GC-
To construct the DNA cassettes, 10 µL of each oligonucleotide (~35 µmol/L) were heated for 5 min to 94 °C. When the temperature reached 40 °C, 10 µL of a reaction mix was added [reaction mix: 2 mmol/L dNTPs; 5 U Klenow enzyme (Invitrogen); 2 µL 10×reaction buffer 2 (Invitrogen); 10% Me2SO; H2O to 10 µL]. All the reactions were performed identically using a thermo-cycler to precisely control the temperatures (Figure 1).
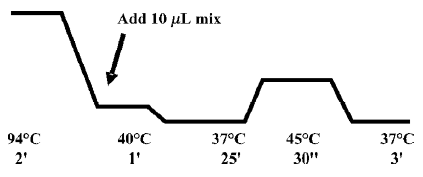
Then, the Klenow products were heated to 65 °C to inactivate the enzyme, digested with HindIII and cloned into the HindIII restriction site of the pBB4H 1 vector previously dephosphorylated with alkaline phosphatase (Roche, Basel Switzerland). Only the correct cassettes would be ligated into the HindIII cohesive ends. We did not observe clones with tandem-repeated cassettes.
The sequences targeting the eGFP mRNA were chosen based on 2 criteria: (i) because they had fewer than 3 G or C repeats to prevent G-quartet formation; and (ii) because they were distributed along the eGFP mRNA.
Cell culture and transfection HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and antibiotics, and were seeded every 3 d following standard procedures at 37 °C. The day before transfection, cells were grown without antibiotics in 12 well plates. Vectors containing the DNA cassette were transfected using lipofectamine 2000 (Invitrogen) at different concentrations following the manufacturer’s instructions. The day after transfection, the cells were washed once with phosphate-buffered saline (PBS) and treated with 5 MOI of a recombinant adenovirus encoding the green fluorescent protein (eGFP) gene under the transcriptional control of the cytomegalovirus (CMV) promoter[22]. Four hours later, the cells were washed with PBS and the media was replaced. Twenty-four hours later the fluorescence was evaluated using a FACScan flow cytometer (Becton & Dickinson, Franklin Lakes, NJ). To evaluate the fluorescence intensity, 10 000 events were counted in at least 3 independent experiments for each concentration.
Confocal microscopy The HeLa cells that were treated with the plasmids and adenovirus as described were washed with PBS, trypsinized and plated on sterile coverslips. Twenty-four hours later, the cells were analyzed with a confocal microscope (Bio-Rad, Hercules, CA) using a 40× objective. The confocal images were analyzed using the Confocal Assistant freeware software.
Results
Construction of DNA cassettes For the construction of the DNA sequence that served as a template for shRNA transcription, we followed the principle of DNA amplification commonly used in PCR reactions (Figure 2A). That is, any given sequence that serves as a primer could be amplified at its 3' end with a proper DNA polymerase. In RNAi hairpins, the 2 strands that form the stem are fully comple-mentary, so it is possible to use only a single oligonucleotide that is self-complementary within its 3' end (Figure 2B). After the self-complementary structure is formed, it is possible to further amplify the structure so that the oligonucleo-tide becomes completely double-stranded DNA.
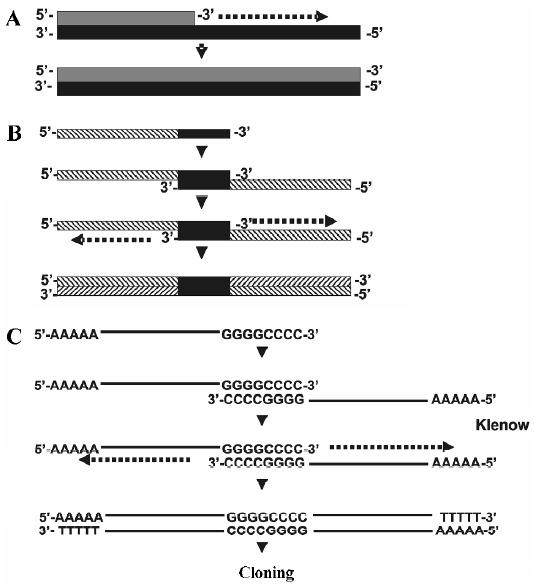
The self-complementary pairing must be strong enough to allow the DNA polymerase to initiate the reaction at the selected temperature. We chose the sequence 5'-GGGGCCCC-3' due to its high Tm (37 °C) and because its palindrome nature permits the self-complementary structure. Furthermore, this sequence allows amplification using the large (Klenow) fragment of the DNA polymerase, whose working temperature is 37 °C. Thus, because the 5'-GGGGCCCC-3' sequence is amplified and lies between the sense and antisense strand, it also functions as a loop for the shRNA (Figure 4).
Finally, most RNAi vectors contain an RNA pol III promoter such as U6 or H1, which is used to initiate the transcription of the shRNA. When the RNA pol III finds 5 consecutive T’s, it terminates the transcription and removes the last three nucleotides on the 3' end[17,20]. Therefore, we included 5 A’s in the 5' end in the oligonucleotide; when the Klenow fragment synthesizes the antisense strand, it generated the 5 T’s, which will be needed to terminate the transcription by RNA pol III inside the cell (Figure 2C). The expected shRNA structures that will form inside the cells are shown in Figure 4. All these structures were modeled using the Mfold server (see Materials and methods).
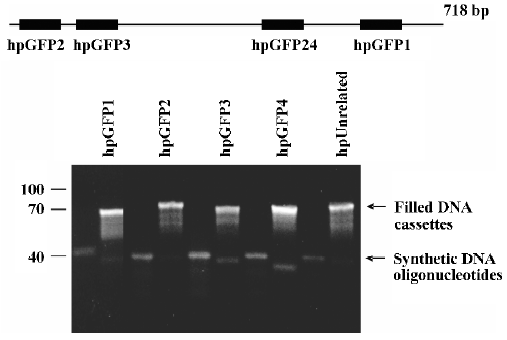
To determine if it was possible to construct the DNA cassette for shRNA vectors with this method, we designed and tested its ability to amplify different oligonucleotides. These oligonucleotides were prepared to target sequences from eGFP and a control oligonucletide containing an eGFP-unrelated sequence. eGFP oligonucleotide sequences were selected to have low GC repeats to prevent G-quartet forma-tion. Figure 3 illustrates the result of a typical filling-in reaction, showing that the Klenow fragment produced double-stranded DNA from single oligonucleotides. Therefore, the self-complementary 5'-GGGGCCCC-3' sequ-ence is a good template for DNA amplification. Furthermore, all cassettes were constructed simultaneously in a single step reaction, so the 1-oligonucleotide method could be automated to perform a large number of reactions in a short period of time (see Materials and methods and Figure 1). We cloned the DNA cassettes in the vector under the transcriptional control of an RNA pol III (H1) promoter, and all cassettes were sequenced to confirm their integrity.
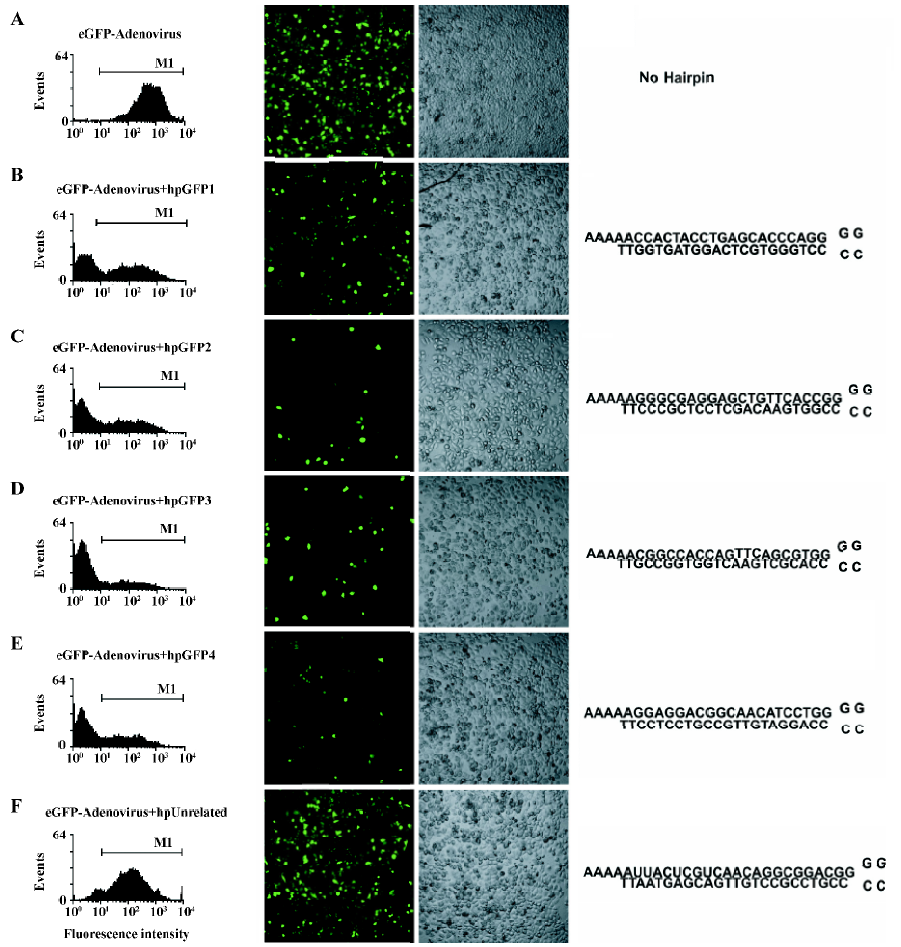
Efficacy of the 1-oligonucleotide method in RNAi experiments To test the ability of the DNA cassettes to trigger RNAi activity, HeLa cells were transfected with the plasmid vectors containing the cassettes targeting the mRNA from eGFP and a control construct. The day after transfection, cells were incubated with a recombinant adenovirus encoding the eGFP, and 24 h later the cell fluorescence was evaluated. All the tested sequences silenced eGFP expression and, as expected, there was no gene silencing with the transfection of the unrelated construct (Figure 4). A dose–response experiment showed that the most effective plasmid concentration to achieve RNAi was 1−3.75 µg/mL. Of 4 tested DNA cassette sequences (termed hpGFP1 to 4), hpGFP 2, 3 and 4 inhibited eGFP expression/fluorescence by more than 70%. The less efficient cassette (hpGFP1) inhibited eGFP expression by 50% at 1 µg/mL (Figure 5). The EC50 of each shRNA confirms this result; the less effective shRNA is hpGFP1, with an EC50 of approximately 0.5 µg/mL, whereas hpGFP 2–4 have EC50 values of approximately 0.05 µg/mL (Figure 5). The finding that not all DNA cassette sequences had the same potency confirms the fact that a variety of sequences must be designed for the same target in order to find the most effective RNAi inducer. However, it was surprising that the hpGFP 1 sequence was the least effective of all tested sequences. This result was interesting because this sequence had been shown to be a strong inhibitor of eGFP expression in a previous study[23]. The dose-response experiment with the unrelated cassette did not influence the eGFP fluorescence (data not shown). Together, these results suggest that diverse DNA cassettes must be produced by the 1-oligonucleotide method in order to identify the best sequence for RNAi.
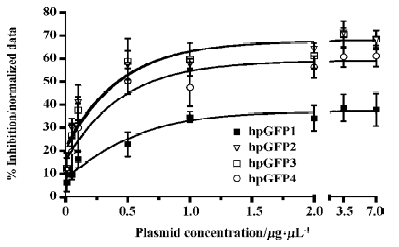
Discussion
In the present work we developed a novel, easy and efficient 1-oligonucleotide method to generate DNA cassettes for RNAi vectors. To test the capability of the method, we designed DNA cassettes to target the mRNA from eGFP. For this purpose a recombinant adenovirus encoding the eGFP gene was used to infect HeLa cells. Only cells infected with the virus and transfected with specific anti-GFP cassettes strongly inhibited eGFP fluorescence. The level of inhibition achieved with this method resembles the inhibition when RNAi is used to knock-down the expression of endogenous genes, because transfections usually reached 60%–75% of the cells, and the level of adenovirus infection almost reached 100%. The results obtained here suggest that eGFP expression was strongly reduced in the cells that were transfected with the DNA constructs, as illustrated in Figure 5. The fact that hpGFP1 was less efficient at knocking down eGFP expression suggests that a variety of RNA sequences are needed in order to find the most effective. Although this sequence has already been used by others[23], it is reasonable that there could exist sequences that are even more effective; this method is an easy way to construct a large variety of DNA cassettes for 1 or more genes. Together, these results demonstrate that, although eGFP expression is under the transcriptional control of the strong CMV promoter in the adenoviral vector, it is still possible to induce gene silencing using the cassettes described here.
Previously reported methods to produce RNAi using DNA cassettes to generate shRNAs in mammalian cells require long oligonucleotides or several PCR reactions that are sequence and step specific[17,20,21,24–28]. For example, in the method reported by Gou et al, at least three oligonucleotides (1 forward and 2 reverse) are used in 2 sequential PCR reactions to generate the DNA cassette[21]. Once the 2 PCR reactions conclude, PCR products require further cleaning before transfection into cells. Another method previously reported is based on primer extension, and the DNA cassettes are cleaned and introduced in the cells directly[26]. Although the latter method uses only 2 oligonucleotides to construct each DNA cassette, these oligonucleotides are fairly long (approximately 100 nucleotides), considerably increasing the cost of constructing multiple cassettes.
There are a number of effective methods that have been designed over the past few years to produce RNAi in mammalian cells[29], but unfortunately none of these methods is easily automated because they require many step-specific reactions. Using the method described here, we simultaneously produced a large number of different DNA cassettes targeting different mRNAs (unpublished data). This is possible because the process can be automated to perform many different reactions using the same temperature, time and reactant conditions using a thermocycler.
The advantages of the method described here over previously reported methods are: (i) a single oligonucleotide is sufficient to generate a DNA cassette for shRNA production, significantly reducing the price of testing multiple sequences; (ii) oligonucleotides for cassette construction are easy to design: a 21–25 nucleotide sense target sequence is flanked by 5'-AAAAA- and -GGGGCCCC-3', thus the length of the oligonucleotide is considerably shorter; and (iii) the method is automated, allowing the construction of a wide variety of DNA cassettes in one step in a few minutes.
Recently it was shown that it is also possible to express synthetic miRNAs using RNA pol II promoters, which allows still more control over stem-loop RNA expression, because many RNA pol II promoters function in a inducible/tissue-specific fashion[30]. We are currently constructing DNA cassettes by using the 1-oligonucleotide method for use with RNA pol II promoters (unpublished results). Because this type of polymerase does not terminate the transcription after 5 consecutive thymidines it is not necessary to include 5 adenines in the oligonucleotide, which further reduces the length and price of the DNA cassette.
In conclusion, we reported here a novel method for constructing large libraries of RNAi cassettes quickly and in a cost-effective manner, thus making the exploration of the function of a large number of genes an easier task and facilitating functional genomic studies.
Acknowledgements
We thank Agustin LUZ for providing genomic DNA for cloning the H1 human promoter, and Erendira AVENDAÑO and members of the laboratory for helpful discussions.
References
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000;101:25-33.
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000;404:293-6.
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001;409:363-6.
- Nykanen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 2001;107:309-21.
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004;305:1437-41.
- Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003;115:199-208.
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 2002;16:948-58.
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550-3.
- Alibu VP, Storm L, Haile S, Clayton C, Horn D. A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol Biochem Parasitol 2005;139:75-82.
- Hosono T, Mizuguchi H, Katayama K, Xu ZL, Sakurai F, Ishii-Watabe A, et al. Adenovirus vector-mediated doxycycline-inducible RNA interference. Hum Gene Ther 2004;15:813-9.
- Czauderna F, Santel A, Hinz M, Fechtner M, Durieux B, Fisch G, et al. Inducible shRNA expression for application in a prostate cancer mouse model. Nucleic Acids Res 2003;31:e127.
- Robinson R. RNAi therapeutics: how likely, how soon? PLoS Biol 2004;2:E28.
- Wesley SV, Liu Q, Wielopolska A, Ellacott G, Smith N, Singh S, et al. Custom knock-outs with hairpin RNA-mediated gene silencing. Methods Mol Biol 2003;236:273-86.
- van den Haute C, Eggermont K, Nuttin B, Debyser Z, Baekelandt V. Lentiviral vector-mediated delivery of short hairpin RNA results in persistent knockdown of gene expression in mouse brain. Hum Gene Ther 2003;14:1799-807.
- Liu XD, Ma SM, Liu Y, Liu SZ, Sehon A. Short hairpin RNA and retroviral vector-mediated silencing of p53 in mammalian cells. Biochem Biophys Res Commun 2004;324:1173-8.
- Zhao LJ, Jian H, Zhu H. Specific gene inhibition by adenovirus-mediated expression of small interfering RNA. Gene 2003;316:137-41.
- Xia XG, Zhou H, Ding H. Affar el B, Shi Y, Xu Z. An enhanced U6 promoter for synthesis of short hairpin RNA. Nucleic Acids Res 2003;31:e100.
- Shen C, Buck AK, Liu X, Winkler M, Reske SN. Gene silencing by adenovirus-delivered siRNA. FEBS Lett 2003;539:111-4.
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002;2:243-7.
- Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci USA 2002;99:1443-8.
- Gou D, Jin N, Liu L. Gene silencing in mammalian cells by PCR-based short hairpin RNA. FEBS Lett 2003;548:113-8.
- Tattersfield AS, Croon RJ, Liu YW, Kells AP, Faull RL, Connor B. Neurogenesis in the subventricular zone following transcranial magnetic field stimulation and nigrostriatal lesions. J Neurosci Res 2004;78:16-28.
- Billy E, Brondani V, Zhang H, Muller U, Filipowicz W. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc Natl Acad Sci USA 2001;98:14428-33.
- Castanotto D, Rossi JJ. Construction and transfection of PCR products expressing siRNAs or shRNAs in mammalian cells. Methods Mol Biol 2004;252:509-14.
- Castanotto D, Li H, Rossi JJ. Functional siRNA expression from transfected PCR products. RNA 2002;8:1454-60.
- Sioud M, Leirdal M. Potential design rules and enzymatic synthesis of siRNAs. Methods Mol Biol 2004;252:457-69.
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA 2002;99:6047-52.
- Paddison PJ, Caudy AA, Sachidanandam R, Hannon GJ. Short hairpin activated gene silencing in mammalian cells. Methods Mol Biol 2004;265:85-100.
- Hannon GJ, Conklin DS. RNA interference by short hairpin RNAs expressed in vertebrate cells. Methods Mol Biol 2004;257:255-66.
- Zeng Y, Cai X, Cullen BR. Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol 2005;392:371-80.