
Molecular mechanism and regulation of autophagy1
Introduction
Cell homeostasis is maintained by a precisely regulated balance between synthesis and degradation of cellular components. There are 2 powerful hydrolytic mechanisms in eukaryotic cells: the proteasome and the lysosome/vacuole. More than 90% of cellular proteins are long-lived. These proteins and some cytoplasmic organelles are believed to be degraded within a specific compartment, the lysosome/vacuole[1]. There are at least 3 different pathways for lysosomal protein degradation: Cvt (cytosol to vacuole targeting pathway)[2], Vid (vacuolar import and degradation path-way)[3] and autophagy[4]. Autophagy plays important roles in physiology and pathophysiology in all cell types. In addition to its role in protein and organelle degradation, autophagy may induce a type of programmed cell death that is different from apoptosis, namely type II programmed cell death[5]. Recently it has been found that autophagy may be a transitory tactical response, and that it affects a range of normal developmental processes, such as sporulation in yeast and pupa formation in Drosophila melanogaster[6,7]. Furthermore, autophagy may contribute to the extension of lifespan induced by caloric restriction[8]. Autophagy might also act as a means of defense against invasion by various bacteria and viruses. There is a potential link between autophagy and a number of diseases in humans. For example, cancer, cardiomyopathy and neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases, amyotrophic lateral sclerosis and prion diseases are all associated with increased autophagy activity[9]. Consequently, study of the molecular basis of autophagy would result in a better understanding of the role of autophagy in cell death and ageing, and would also result in proposals for new therapeutic approaches for neurodegenerative diseases.
The morphology of autophagy was first characterized in studies of mammalian cells. With a few exceptions, however, the molecular components of autophagy were initially elucidated in yeast because of the convenience of gene analysis. Recent studies in various eukaryotic systems have revealed a conservation of the autophagic mechanism[10]. In the past, the many terms used in the autophagy field have been highly confusing: Aut (autophagocytosis), Apg (autophagy), Vps (vacuolar protein-sorting) all have been used. Recently, the autophagy-related genes and the products of these genes were named ATG and Atg, respectively[11].
Classification of autophagy
Autophagy is a ubiquitous physiological process that occurs in all eukaryotic cells. There are three primary forms of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Macroautophagy is the most prevalent form of autophagy. It comprises the following processes: initially, a “C” shape double-membrane structure appears in the cytoplasm, and then at both ends of this membrane a structure grows, and finally closes to form a vacuole. The bulk of the cytoplasm and some organelles are wrapped into the vacuole (autophagosome). Then the autophagosome targets the lysosome/vacuole, where its outer membrane fuses with the lysosomal membrane and the inter sac (autophagic body) enters the lysosome/vacuole. The autophagic body is degraded in the lysosome/vacuole so the carrying constituent components can be recycled[12]. Microautophagy is a form with few features. In this pathway, the membrane of the lysosome/vacuole invaginates, and then finally pinches off to form an internal vacuolar vesicle that contains material derived from the cytoplasm, akin to the autophagic bodies formed in macroautophagy. The notable difference between macroautophagy and microautophagy is that in the latter the cytoplasm is directly up taken into the lysosome/vacuole[13]. CMA differs from the other lysosomal degradation pathways in that vesicular traffic is not involved. Cytosolic proteins with particular peptide sequence motifs are recognized by a complex of molecular chaperones, then bind to a receptor in the lysosomal membrane, the lysosome-associated membrane protein (Lamp) type 2a. Proteins are delivered to lysosomes with the help of molecular chaperones and Lamp 2a[14].
Autophagy is basically a non-selective process, in which bulk cytoplasm is randomly sequestered into the cytosolic autophagosome. However, in some cases it may select its target. For example, autophagy can selectively eliminate some organelles, such as injured or excrescent peroxisomes, endoplasmic reticulum (ER) and mitochondria[15]. A recent report shows that in yeast, Saccharomyces cerevisiae, the cytosolic protein acetaldehyde dehydrogenase (Ald6) is delivered to the vacuole and degraded by means of specific autophagy[16].
Molecular mechanisms of macroautophagy
Macroautophagy, a major form of autophagy, is relatively well characterized at present. Autophagy and the related processes are dynamic, and many molecules involved in the autophagic process have been identified. At least 25 specific yeast genes are exclusively involved in autophagy, and there are more than 40 additional yeast genes required for autophagy, but they also play roles in other pathways[11]. However, the physiological functions of many of these genes need to be further clarified.
Origin of the autophagosomal membrane There is still debate on the origin of autophagosome membranes. Initially, the ribosome-free region of the rough ER and Golgi were proposed as the source of autophagosomal membranes[17,18]. Now, it is generally accepted that the phagophore, a poorly characterized organelle, may be the major source of the autophagosomal membrane and related structures[19].
Autophagosome formation Two ubiquitin-like conjugation systems In yeast, autophagy almost completely shuts off under growing conditions, although every ATG gene is expressed. Molecular biological and biochemical analyses of these gene products uncovered the genetic and biophysical interactions among the Atg proteins. One of the most remarkable findings regarding the Atg proteins is the discovery of two ubiquitin-like conjugation systems, Atg12-Atg5 and Atg8- phosphatidylethanolamine (PE) (Figure 1). In fact, half of the APG genes essential for autophagy are involved in these conjugation systems, and these two conjugation systems are well conserved among eukaryotes. Furthermore, Atg12-Atg5 and Atg8 conjugation systems are somehow related to each other[20–22]: if the former is defective, the latter can not target to the pre-autophagosomal structure (PAS); the levels of Atg8-PE also play an important role in the conjugation of Atg12-Atg5.
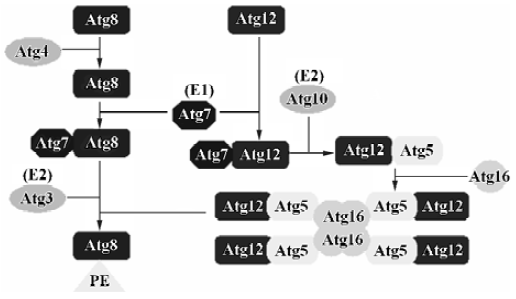
Atg12-Atg5 conjugation system Atg12, a small hydrophilic protein of 186 amino acids with no apparent homology to ubiquitin, can covalently link to a unique target protein, Atg5[23]. The mode of conjugation of Atg12 and Atg5 is quite similar to that of ubiquitination. Atg12 is first activated in an ATP-dependent manner by Atg7 (it functions as an ubiquitin-activating enzyme, E1), leading to the formation of a thioester bond between the C-terminal glycine in Atg12 and a cysteine residue in Atg7[24]. The C-terminal glycine in Atg12 is then transferred to the cysteine in Atg10 (it functions as a ubiquitin-conjugating enzyme, E2), forming a new thioester bond, and Atg7 is released[25]. Finally, the C-terminal glycine in Atg12 forms an isopeptide bond with the ε-amino group of lysine 149 in Atg5, and Atg10 is in its free state again. The formation of the Atg12-Atg5 conjugate is indispensable to autophagosome formation. It seems that this ubiquitin-like system is a constitutive process, because the formation of the Atg12-Atg5 conjugate is not dependent upon starvation or other autophagy-inducing conditions[26]. Atg12 and Atg5 form a conjugate immediately after their synthesis, and free forms of these are hardly detectable. The conjugation reaction between Atg5 and Atg12 is irreversible, and so far no protease has been found to deconjugate this conjugate. Atg16 also binds preferentially to the Atg12-Atg5 conjugate. Atg16 links with Atg12-Atg5 through self-oligomerization, and its C-terminal coiled-coil region may be responsible for this oligomerization. Therefore, Atg16 forms a 350 kDa multimeric complex with the Atg12-Atg5 conjugates. It should be pointed out that Atg16 only binds to Atg5, and not to Atg12[27,28]. This new complex is necessary for the elongation of the isolation membranes (used for formation of the autophagosomal membrane). A small fraction of the Atg12-Atg5·Atg16 complex initially associates with a small crescent-shaped vesicle evenly. As the membrane elongates, Atg12-Atg5 shows asymmetric localization and most of these proteins associate with the convex surface of the isolation membrane. This complex will dissociate from the membrane upon completion of autophago-some formation; thus, it is not present in the mature autophagosome[29]. The molecular basis of this transient association of Atp12-Atg5 conjugates with the autophago-some membrane is not yet known.
In mammalian cells, Atg5 and Atg12 are conjugated to each other in the same way as they are in yeast, but the complex interacts with Atg16L, forming an ~800 kDa structure instead of a 350 kDa complex in yeast[27]. Atg16L is a 63 to 74 kDa protein, which has a binding region and coiled-coil region similar to that of Atg16. However, Atg16L has a long C-terminal extension containing 7 WD repeats, but the role of the WD repeats in autophagy has not yet been elucidated.
Atg8 conjugation system The second ubiquitin-like protein essential for autophagy is Atg8 (Aut7/Apg8). Atg8 is a 117-amino acid protein and is present in the early isolation membranes, autophagosomes and autophagic bodies[30]. This feature makes Atg8 a good marker for studying membrane dynamics during autophagy. Cell fractionation studies have shown that Atg8 is mostly membrane-bound; approximately half of it is peripherally bound to the membrane and the other half behaves like an intrinsic membrane protein. Atg4, a novel cysteine protease, is responsible for processing Atg8 by cleaving a single Arg residue from it, consequently exposing Gly in the C-terminus of Atg8[31]. Atg8 can be activated by Atg7 (E1) in an ATP-dependent manner and transferred to a conjugating E2 enzyme, Atg3[32]. Atg7 is a unique enzyme that activates two different ubiquitin-like proteins, Atg12 and Atg8, and assigns them to their proper E2 enzymes, Atg10 and Atg3, respectively. Interestingly, in the final step, Atg8 does not form a conjugate with other proteins, but interacts with PE, an abundant membrane phospholipid[32]. This lipidation reaction leads to a conformational change of Atg8 that is necessary for the membrane dynamics of autophagy[33]. In addition, Atg8-PE is deconjugated by Atg4, which cleaves the lipid-protein linkage and provides a new source of cytoplasmic Atg8. The cycle of conjugation and deconjugation is important for the normal progression of autophagy.
Microtubule-associated protein 1 light chain 3 (LC3), the mammalian orthologue of Atg8, targets to the autophago-somal membranes in an Atg5-dependent manner and remains there even after Atg12-Atg5 dissociates. Thus LC3 is the only credible marker of the autophagosome in mammalian cells[12]. In wild-type cells, LC3 is detected in 2 forms: LC3-I (18 kDa) and LC3-II (16 kDa)[34]. Twenty-two amino acids in the C-terminus of the newly synthesized LC3 are cleaved immediately by the mammalian orthologue of the yeast cysteine proteinase Atg4, autophagin, to produce an active cytosolic form, LC3-I[35]. Then with the catalysis of Atg7 and Atg3, LC3-I undergoes a series of ubiquitination-like reactions, and is modified to LC3-II. LC3-I is located in the cytoplasm, while LC3-II is a tightly membrane bound protein and is attached to PAS and autophagosomes. The relative amount of membrane-bound LC3-II reflects the abundance of autophagosomes, so the induction and inhibition of autophagy can be monitored through measuring total and free LC3-II levels by means of immunoassay[34]. In addition, studies have shown that the Atg12 and LC3 systems have a functional relationship. In ATG5–/– cells, LC3-II is not generated at all[21]. As a result, LC3 cannot target the autophagosomal membranes. The recent generation of transgenic mice expressing green fluorescent protein (GFP) fused to LC3 provides a useful tool to investigate autophagy in various mammalian organs in vivo[36].
In addition to LC3, at least another two mammalian orthologs of yeast Atg8 have been identified[37,38]: γ-aminobutyric acid type A receptor-associated protein (GABARAP) and Golgi-associated ATPase enhancer of 16 kDa (GATE-16). The 2 proteins also covert to membrane bound forms (form II), which are recovered in membrane fractions[39]. These results suggest that all mammalian Atg8 homologues receive common modifications to associate with autophagosomal membranes, but the functions of these orthologs and their modified form II need to be further studied.
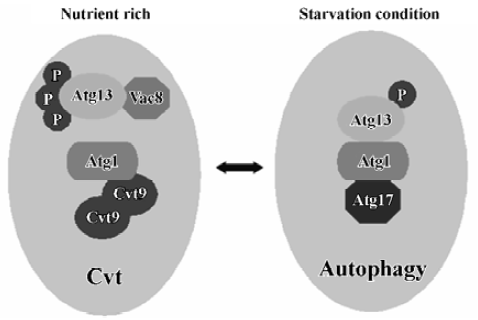
Two kinase complexes Atg 1 protein kinase Atg1 is a serine/threonine protein kinase, which forms a protein complex with different regulatory proteins such as Atg13, Vac8, Atg17 and Cvt9. The complex controls the switch between Cvt and autophagy pathways (Figure 2). The composition of this complex is dynamic and it may vary depending on nutrient condi-tions[40]. Under nutrient-rich conditions, Atg13 is hyperphos-phorylated so that its association with Atg1 is blocked. On the other hand, under nutrient starvation conditions or after treatment with rapamycin, Atg13 becomes partially dephos-phorylated. This dephosphorylation leads to an Atg1-Atg13 interaction and subsequent generation of autophagosomes instead of Cvt vesicles and, thus, activates autophagy. Vac8, the vacuolar inheritance protein that acts in the Cvt pathway is not essential for starvation-induced autophagy[41]. Vac8 is also a phosphoprotein and may help to facilitate the phosphorylation of Atg13. Atg1 also interacts with 2 other proteins. One of these is Cvt9, which is only required for the Cvt pathway[42]. Cvt9 is a large coiled-coil protein, and it interacts with itself, possibly through the coiled-coil domain. This could potentially crosslink the Atg1 complex into a higher-order structure required early in the sequestration process of autophagy. The other protein, Atg17, is only required for the autophagic import and has been proposed to play a role in Atg1-Atg13 interactions[43]. Recently, Atg17 has been found to interact with 2 Cvt pathway-specific components, Atg24/Cvt13/Snx4 and Atg20/Cvt20, proteins that contain PI3P-binding PX domains[44,45]. But it remains a challenge to evaluate the meaning of interactions between the autophagic-specific Atg17 protein and Cvt pathway-specific Atg24 and Atg20 proteins. Genetic analysis suggests that the Atg1 complex functions at a rather late stage of autophagosome formation[22]. The Atg1 complex may control membrane dynamics, rather than act as a signal transducer. The kinase activity of Atg1 is upregulated during induction of autophagy, thus the levels of kinase activity seem to be important for the regulation of autophago-some formation[43]. A recent report has shown that Atg1 may only have a non-kinase structural role in autophagy induction, although the kinase activity of Atg1 is involved in autophagy; however, it plays more important roles in the Cvt pathway[46].
Two putative human homologs of Atg1 have been identified[47]: the UNC-51-like kinases ULK1 and ULK2. ULK1 has been shown by yeast two-hybrid screening to interact with GATE-16 and GABARAP, two homologs of the yeast autophagy protein Atg8.
Vps34/PI3K Early in 1982, it was found that 3-MA (3-methyladenine) inhibits the formation of autophago-somes[48]. 3-MA is a PI3K inhibitor[49]. Several studies have demonstrated that PI3K inhibitors interfere with the formation of autophagosomes in rat hepatocytes[50]. Yeast is known to contain only one PI3K, Vps34[51]. Vps34 function is regulated by the protein kinase activity of Vps15, with which it forms a stable, membrane-associated complex under normal conditions. This complex links the Vps34 kinase to cytoplasmic membranes. Vps34-Vps15 is present in 2 complexes, which are involved in a variety of membrane transport events (Figure 3)[52]: complex I, which is composed of Vps34-Vps15, Atg6 and Atg14, controls autophagy, whereas complex II, which is composed of Vps34-Vps15, Atg6 and Vps38, is essential for sorting of carboxypeptidase Y (CPY) into the vacuole. Atg6 is a possible coiled-coil protein and is associated with the membrane through Vps15 and Vps34. Atg14 is a specific factor in the autophagy-specific PI3K complex. Complex I functions primarily, but not exclusively, at the PAS, whereas complex II functions at the endosome. The lipid kinase activity associated with Vps34 is thought to create lipid patches of PI3-P, the reaction product of class III PI3K, at specific trans-Golgi locations, and these patches then function in protein sorting into vesicles that travel from the Golgi to the endosome.
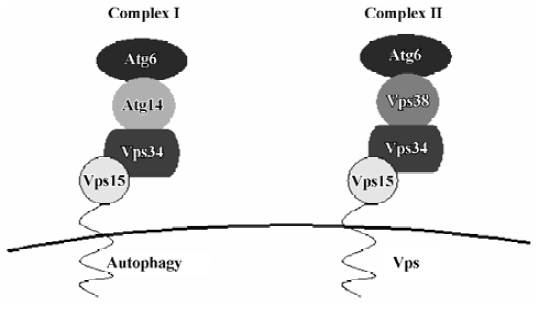
In mammalian cells, there are 3 classes of PI3K. Class I PI3K is an inhibitor of autophagy[53]. Class II PI3K activity is thought to have no relevance to autophagy control. Class III PI3K, a functional orthologue of yeast Vps34, is an activator of autophagy and plays a crucial role at an early step of autophagosome formation in mammalian cells[54], and it is required for increasing the size of the sequestering membrane, presumably through fusion events. PI3-P interacts with proteins containing FYVE or PX motifs, thus recruiting such proteins from the cytosol for autophagosome biogenesis[55]. The activation of a population of PI3K located in a determined membrane domain may be responsible for autophagosome biogenesis. In addition, the presence of PI3-P in a specific membrane location may generate significant asymmetries and drive membrane curvature of PAS[54]. Finally, PI3-P may be converted to higher-order polyphos-phoinositides (PI), which are involved in diverse signaling functions. The mammalian cell orthologue of Vps15 is p150. It is associated with class III PI3K, and interacts with beclin-1, a functional orthologue of yeast Atg6 in mammalian cells[56]. Beclin1, which is the first autophagy-related tumor suppressor gene reported so far, is required for both autophagy and Cvt pathways. The characterization of the tumor suppressor activity of beclin-1 could establish an important relationship between cancer and the autophagic pathway. Beclin-1 was originally isolated as a bcl-2-interacting protein that can downregulate bcl-2, but it is still unclear whether this interaction is instrumental in autophagy[57].
Atg 9 complex Atg9 is an integral membrane protein, containing several potential transmembrane domains[58]. A fraction of Atg9 is located in the PAS together with other autophagy proteins, but it is absent in the membranes of mature autophagosomes[59,60]. If cells lack Atg9, neither the Cvt pathway nor autophagy can take place. However, Cvt and autophagy can still accumulate protease-sensitive prApe1 (the precursor to aminopeptidase I), indicating that Atg9 has an effect prior to closure of the double-bilayer vesicle[61]. A soluble protein, Atg2 physically interacts with Apg9, and this interaction is indispensable to autophago-some formation[62]. The localization of Atg2 to the PAS requires the activity of several proteins, such as Atg1 and Atg9, but the kinase activity of Atg1 seems to be of questionable importance to this function. Atg18 is also indispensable to correct Atg2 localization, suggesting that there is a potential link between this protein and the Atg9 system[63].
Mammalian orthologs of Atg2 and Atg9 are present in the genome sequence databases, but they have not been studied in detail.
PAS All the above mentioned Atg proteins have a function before or during the formation of autophagosomes. Thus, it is important to find out the distribution of these Atg proteins in cytoplasm. By using fluorescent protein fused with Atg proteins, it has been shown that Atg8 stains autophagosomes, autophagic bodies in the vacuoles, and also the intermediate isolation membrane. However, Atg5 shows a single bright dot structure next to the vacuole. This structure has been named the PAS, and Atg8 also co-localizes with it[64,22]. In addition, no Golgi, ER or late-endosomal markers has been found in the PAS, suggesting that this is a novel structure that has not been described so far[59]. However, little is known about the existence of a PAS-like structure in mammalian cells. Almost all Atg proteins are co-localized in the PAS, suggesting that it is an organizing center of the autophagosome. The autophagy-specific PI3K complex, PI3-P, may recruit different proteins such as Atg18, Atg20, Atg21, Atg24, and Atg27 to the PAS[54,65,66]. The interaction between PI3-P and these proteins is a requisite for the re-recruitment of Atg proteins to the PAS. None of these proteins contains PX or FYVE motifs, so the domain of the lipdation has not been identified. Atg9 also has a strong effect on organization of the PAS, whereas defects in the Atg1 kinase complex show little effect on the PAS structure. Indeed, a recent study suggests that, in nutrient-rich conditions, the PAS does not form in the absence of prApe1, Atg19 or Atg11[67].
Autophagosome fusion with the vacuole/lysosome In yeast, the fusion of autophagosomes with the vacuole requires several factors that are involved in other types of vesicular transport. Molecular genetic studies have indicated that the machinery required for homotypic vacuole fusion is also required for the fusion of autophagosomes with the vacuole. This machinery includes: the SNARE proteins Vam3, Vam7, Vti1, and Ykt6; the NSF, SNAP, and GDI homologs Sec17, Sec18, and Sec19; the Rab protein Ypt7; members of the class C Vps/HOPS complex; and Ccz1 and Mon1[68]. However, it remains to be determined whether different SNARE and/or other fusion components operate during autophagy. If the fusion process begins prior to completion of the double-membrane vesicle, the cargo will remain in the cytosol, so the timing of vesicle fusion with the lysosome/vacuole must be precisely regulated. Atg8-PE, which is located on the outer surface of the autophagosome, is removed prior to fusion as a result of a second cleavage by Atg4. Removal of Atg8-PE from autophagosomes can prevent premature fusion with lysosomes. Atg12-Atg5•Atg16 may have the same function. The expression of Atg8ΔR is a mutated form of Atg8 lacking the ultimate arginine residue, bypassing the need for the initial Atg4-dependent cleavage step. However, the mutation causes a loss of ability of Atg to carry out the second cleavage event that releases Atg8 from PE and results in a partial defect in autophagy[31].
In mammalian cells, autophagosome fusion with lysosomes is a more complex process in which the autophagosome requires a series of maturation steps prior to its fusion with the lysosome. Similar to yeast, the activity of monomeric GTPases (Rab22, Rab24) and mammalian orthologs of SNARE protein family members and the NSF protein are needed for correct autophagosome maturation[69,70]. Prior to their fusion with lysosomes, autophagosomes have to fuse with endosomes or endosome-derived vesicles. Overexpres-sion of a mutant form of the regulator of endosome sorting, SKD1 ATPase, which is unable to hydrolyze ATP, hampers endosome function and causes a massive accumulation of nascent autophagosomes[71]. Rab7, which is associated with autophagic vacuoles, is involved in maturation of autophago-somes. Overexpression of a Rab7 dominant negative mutant hampers fusion between autophagosomes and the late endosome/lysosomal compartment, leading to the accumulation of autophagosomes with a concomitant decrease in the degradation of long-lived proteins[72]. In addition, the cyto-skeletal elements are also involved in either autophagosome maturation or autophagosome-lysosome fusion. The microtubule is another important factor for this event, because treatment of cells with microtubule-destabilizing drugs blocks autophagosome maturation. For example, cells treated with cytochalasin D, an agent that disrupts actin filaments, display a significant reduction in autophagosome formation[73], whereas the microtubule stabilization mediated by a new antitumor drug, taxol, increases the fusion of amphisomes with lysosomes[74].
Degradation of autophagic body The main purpose of autophagy in yeast is to degrade cytoplasm and recycle the resulting macromolecules for use in the synthesis of essential components during nutrient stress. Degradation of autophagy bodies requires a low pH, proteinase B (Prb1) and Atg15/Cvt17[75,76]. Prb1 is a hydrolase that is involved in the activation of many other vacuolar zymogens, which indirectly affects the vesicle breakdown. Atg15/Cvt17 has sequence similarity to a family of lipases, and seems to function directly in vesicle breakdown. It is the only putative lipase that has been identified that has a role in the degradation of autophagic bodies[76]. So far, we know that the expression of Atg15 is low in the vesicle, but the mechanism or site of action of Atg15 is unknown. Another protein, Atg22, has also been implicated in this last step. Atg22 is an integral membrane protein, and is needed only for the degradation of autophagic bodies[77].
Molecular mechanisms of CMA
CMA is one of several lysosomal pathways of proteo-lysis. CMA is activated by physiological stressors such as prolonged starvation. During starvation, macroautophagy is activated first, but this process quickly declines and CMA is then activated. The mechanisms by which these 2 lysosomal protein degradation pathways interact, if any, are unknown. We now know that cytosolic proteins can be degraded by CMA in rat liver, kidney, heart, and other tissues. However, to date, there are no physiological phenomena similar to CMA found in yeast. Only the cytosolic proteins with exposed pentapeptide sequence motifs related to KFERQ can act as substrate proteins, and be recognized by a complex of molecular chaperones whose major constitutive form is the heat shock 70 kDa protein (hsc70)[78]. The substrate protein and molecular chaperone complex bind to the lysosomal membrane and interact with lamp2a. The protein is unfolded by the molecular chaperone complex prior to importation into the lysosome. The protein is pulled into the lysosomal lumen with the help of lysosomal hsc70 (ly-hsc70)[14,79]. The level of lamp2a can be a rate-limiting step in CMA. A lamp2 knockout mouse without any of the lamp2 isoforms showed a reduction in rate of protein degradation and an increase in accumulation of autophagic vacuoles in heart, skeletal muscle, and other tissues[80]. The amount of lamp2a in the lysosomal membrane is regulated, in part, by changes in its degradation rate. For example, during starvation lamp2a levels increase due to a decrease in lamp2a degradation. The mechanisms of lamp2a degradation include an initial cleavage by the lysosomal protease cathepsin A. In fact, cathepsin A knockout mice had elevated levels of lamp2a in the lysosomal membrane and showed higher activity of CMA[81]. A metalloprotease yet to be identified also contributes to lamp2a degradation and works in cooperation with cathepsin A. A more rapid adjustment of lamp2a in the lysosomal membrane occurs through changing the proportions of lamp2a sequestered in the lysosomal matrix.
Regulation of autophagy
Autophagy is probably the main mechanism for degradation of long-lived proteins and cytoplasmic organelles. A great number of extracellular stimuli (starvation, hormone or therapeutic treatment) as well as intracellular stimuli (accumulation of misfolded proteins, invasion of microorganisms) are able to modulate the autophagic response. In yeast and mammalian cells, autophagy is a fundamental biological event that occurs under normal growth conditions. Thus, there must be an array of mechanisms by which extracellular and/or intracellular signals can be accepted and transmitted to the regulatory factors to promote or inhibit autophagy when it is needed.
TOR/mTOR There are a number of signaling complexes and pathways involved in the initiation and maturation of autophagy. The central player in these signaling pathways is TOR, the target of rapamycin. TOR is a serine/threonine kinase involved in most regulatory pathways that control the response to changes in nutrient conditions and energy metabolism. TOR acts as a good gate-keeper in autophagy and exerts an inhibitory effect on autophagy (Figure 4). TOR kinase may inhibit autophagy through two general mechanisms. First, TOR acts in a signal transduction cascade through various downstream effectors to control both translation and transcription[82]. Second, TOR appears to directly or indirectly affect the Atg proteins, resulting in interference with the formation of autophagosomes[83]. In mammalian cells, there is a TOR orthologue, mTOR, which appears to modulate autophagy in a manner similar to that observed in yeast.
Protein phosphatase 2A (PP2A) In yeast, TOR phosphorylates the protein Tap42, causing its association with PP2A[84]. This interaction significantly reduces the enzyme activity of PP2A. Inhibition of TOR by nutrient stress or rapamycin results in the dephosphorylation and dissociation of Tap42 from PP2A. PP2A may then dephosphorylate its targets, which eventually leads to a variety of antiprolifera-tive responses and induction of autophagy. PP2A is a phosphatase that acts on several TOR substrates, including glutaminase (Gln3). Dephosphorylation of Gln3 by PP2A promotes its dissociation from urease 2 (Ure2) and its successive translocation into the nucleus, where it activates the transcription of several genes[85,86]. Some of those genes are parts of the autophagy machinery such as Atg8 and Atg14[87,88].
4E-BP1 mTOR has a serine/threonine kinase activity, and eukaryotic initiation factor 4E binding protein-1 (4E-BP1) is one of its substrates[89]. 4E-BP1 is an inhibitor of translation and can be directly phosphorylated by mTOR. After phosphorylation, 4E-BP1 will dissociate from eukaryotic initiation factor 4E (eIF4E). Free eIF4E binds to the 5' terminal cap structure of RNAs and promotes the progress of translation.
p70S6 kinase p70S6 kinase (p70S6) is also a candidate of the substrates of mTOR, which can be called S6K for short. S6K is a protein kinase of ribosomal 40S subunit S6. Phosphorylation of S6 upregulates the translation of mRNAs containing 5' terminal oligopyrimidine tract (5' Top). The 5' Top mRNAs account for approximately 20% of all cell mRNAs and are foundations of protein biological synthesis. The major products of 5' Top mRNAs include ribosomal protein, elongation factor (EFla, EF2), and polyA binding protein. When nutrition is sufficient, TOR is turned on and the activity of the enzyme S6K increases. Recently, it has been found that S6K is needed for the entire process of autophagy activation, and it must be activated first for maximal activation of autophagy[90]. However, there is a contradictory phenomenon, in that TOR, an activator of S6K, is an inhibitor of autophagy. The possible explanation is that S6K needs to be active under starvation conditions in which its activator, TOR, is turned off. Perhaps, after TOR is switched off, any active S6K remains active for some time, ensuring maximal autophagy induction can be achieved, but other mechanisms might then gradually deactivate the p70S6 kinase, thereby preventing excessive autophagy, which could be harmful.
Hormones The hormones glucagon and ecdysone in Drosophila larvae inhibit TOR by downregulating PI3K-I, resulting in an increase in autophagy. Conversely, the hormone insulin appears to have an inhibitory effect on the autophagic pathway[91,92]. In response to food, insulin is produced; insulin binds a receptor on the surface of cells and triggers a signaling cascade. Insulin first activates its tyrosine kinase receptors, causing these receptors to phosphorylate themselves. P85 as a regulatory subunit of PI3K activates PI3K by association with these phosphorylated insulin receptors. The active PI3K transfers to the inter surface of the cell membrane and phosphorylates the lipid phosphatidylinositol, leading to the activation of protein kinase B (AKT/PKB) and other enzymes. This is followed by the activation of TOR. The latter then negatively regulates Atg proteins to prevent autophagy activation.
Amino acids Amino acids, which are the final products of autophagic protein degradation, act as negative feedback regulators for the process. In almost all cell types, especially in hepatocytes, a combination of leucine and a few other amino acids is very effective in inhibiting autophagy[93–95]. It was discovered that the addition of amino acids (leucine in particular) in the absence of insulin or other growth factors resulted in a strong and fairly rapid stimulation of the phosphorylation of ribosomal protein S6K[96,97]. Phosphorylation of 4E-BP1, similar to that of S6K, requires the presence of amino acids, and amino acids alone, but not insulin alone, stimulate phosphorylation of this protein in a rapamycin-sensitive manner[98,99]. Furthermore, leucine activates glutamate dehydrogenase, which contributes to the ability of leucine to potentiate insulin production in β-cells[100]. Amino acids and insulin act synergistically on both processes to inhibit autophagy[96,97]. The amino acid/TOR signaling pathway can be confronted by the AMP-dependent protein kinase (AMPK). However, this effect can be blocked by rapamycin and two inhibitors of PI3K, wortmannin and LY294002[101]. Recently, it has been found that amino acids can modulate activation of the kinase Raf-1. Raf-1 acts upstream of the mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (Erk1/2) cascades[102]. However, the mechanism by which amino acids regulate the activation of Raf-1 remains to be elucidated.
ATP Autophagic sequestration is ATP-dependent, and the depletion of ATP inhibits autophagic sequestration[103]. Because of adenylate kinase equilibrium in the cell, a fall in ATP is often associated with an increase in AMP. AMPK, which serves as a general integrator of metabolic responses to changes in energy availability, is activated in response to elevations of the AMP/ATP ratio. Thus, autophagy can be suppressed by AMP through activation of AMPK. In addition, high levels of AMP can be reached under hypoxia and other conditions of energy depletion, which also suppress autophagy[104,105]. Different studies suggest that in liver and muscle cells AMPK negatively modulates protein synthesis by impairing the mTOR-dependent signals to p70S6 kinase and 4E-BP1[106], interfering with the occurrence of autophagy. Although AMPK is involved in the control of mTOR signaling, its role in autophagy needs to be clarified. However, Snf1, the yeast homologue of AMPK, has been identified as a positive regulator of autophagy[107].
Atg proteins TOR signaling negatively regulates the association between Atg1 and Atg13. Under nutrient-rich conditions, active TOR causes hyperphosphorylation of Atg13, preventing or modulating its association with Atg1[43]. It is not clear whether TOR directly phosphorylates Atg13. TOR inactivation by starvation or rapamycin treatment promotes the rapid dephosphorylation of Atg13, a process that seems to be independent of PP2A. Dephosphorylated Atg13 binds to Atg1. This association promotes autophosphoryla-tion and activation of Atg1, leading to the induction of autophagy. The inhibition of TOR signaling also promotes the assembling of other regulatory proteins in the membrane of PAS due to increased Atg1 kinase activity. In other words, inactivation of TOR is necessary for the prolongation of pre-autophagosomal membranes and enhancement of the expression of autophagic specific genes such as Atg8.
PI3K-I/PKB The PI3K-I/PKB pathway is involved in the negative modulation of autophagy (Figure 4). If PI3K-I is activated, it will phosphorylate PI4P and PI(4,5)P2 to produce PI(3,4)P2 and PI(3,4,5)P3[108]. These lipids have a role in the PAS structure to recruit proteins necessary for the biogenesis of autophagosomes. These lipids bind to protein kinase B (Akt/PKB) and its activator phosphoinositide-dependent kinase-1 (PDK1) via its pleckstrin homology (PH) domains. Upon lipid binding, Akt/PKB is then activated[108]. Furthermore, PDK1 phosphorylates other kinases, including p70S6 kinase, making them acquire kinase activity[109]. Activation of this pathway by expression of a constitutive active form of PDK1 and PKB has an inhibitory effect on autophagy. The phosphatase PTEN, which selectively hydrolyzes PI(3,4,5)P3, has a stimulatory effect on autophagy by relieving class I PI3K/PKB inhibition[55]. Rapamycin can reverse most of the inhibition of autophagy because of activation of the class I PI3K pathway, which suggests that mTOR is a downstream target. The activation of PI3K/PKB has been shown to relieve the inhibitory effects of the tuberous sclerosis complex (TSC1/TSC2, hamartin/tuberin) on mTOR/p70S6 kinase signaling[110]. TSC2 has a GTPase-activating activity towards monomeric Rheb, which controls mTOR/p70S6 kinase signaling[111], so TSC2 can promote the conversion of Rheb from the GTP-bound state (inhibitor of TOR) to the GDP-bound state (activator of TOR).
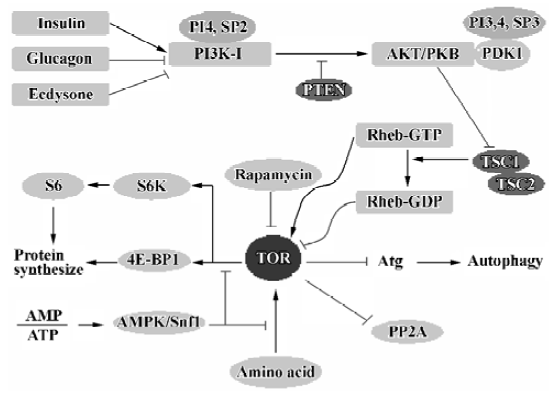
Beclin1/PI3K- III The beclin 1/PI3K-III complex is involved in the formation of autophagosomes and initiation of autophagy. 3-MA, wortmannin, and LY294002, three PI3K-III inhibitors, interfere with this pathway[50]. Recently, it has been shown that in muscle cells, amino acids can negatively regulate autophagy by interference with the activity of class III PI3K[55]. Further studies showed that all beclin forms a complex with PI3K, whereas ~50% of PI3K remains free from beclin. Indirect immunofluorescence microscopy demonstrated that the majority of beclin and PI3K localized to the trans-Golgi network (TGN). Some PI3K also distributed in the late endosome[112]. This suggests that beclin and PI3K control autophagy by functioning in PI3-P sorting into vesicles that travel from the Golgi to the endosome as a complex at the TGN. In addition, an increase in the class III PI3K product, PI3-P, can also stimulate autophagy[49]. Beclin-1, as an important element of mammalian autophagy, is consistently mono-allellically deleted in 40–75% of human sporadic breast, prostate and ovarian cancers. In in vitro cultured MCF-7 cells, overexpression of beclin-1 induces autophagy and is associated with inhibition of MCF-7 cell proliferation[56]. The protein beclin-1 is able to shuttle between the nucleus and the cytoplasm[113]. Its role in the nucleus is unknown, but nuclear beclin-1 does not control autophagy and does not inhibit tumorigenicity of breast carcinoma cells. Beclin-1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor functions[113]. This nuclear export traffic is modulated by nuclear export protein chromosome region maintenance 1(CRM1)[113]. The CRM1-dependent nuclear export traffic plays an important role in the regulation of autophagy. However, when the traffic is inhibited by daunomycin B or a mutation of the nuclear export signal, beclin-1 exists almost exclusively in the nucleus. Thus, autophagy induction by starvation cannot be initiated and its anti-tumor effect is also blunted.
GTPases
Heterotrimeric G proteins and partners In the human colon cancer cell line HT-29, it was found that the trimeric Gi3 (αi3βγ) control the autophagic pathway at the sequestration step[114]. The activity of autophagy was low when the Gαi3 protein was in the GTP-bound form, and it was stimulated when GDP was bound to the Gαi3 protein[115]. The localization of the Gai3 protein is also essential in controlling autophagy. It must first associate with the ER or Golgi in order to control autophagy. The guanine nucleotide cycle of the Gαi3 protein is dependent upon the activity of the G alpha interacting protein (GAIP). GAIP belongs to the regulators of the G-protein signaling (RGS) family and is a GTPase-activating protein towards the Gαi3 protein. The phosphorylation of a conserved serine residue in the RGS domain of GAIP stimulates its GAP activity, and consequently the autophagic pathway[116]. In addition, GAIP is a cytoplasmic substrate for the MAP kinase Erk1/2 and its phosphorylation is reduced in the presence of amino acids. The different domains of the activator of G-protein signaling 3 (AGS3) are all involved in the regulation of autophagy[117]: its N-terminal part containing 7 tetratricopeptide regulatory (TPR) repeats can decrease the occurrence of autophagy; its C-terminal part containing G-protein regulatory (GPR or GoLoco) motifs can interact and stabilize the Gαi3 protein in its GDP-bound conformation. Recently, investigators showed that AGS3 had a stimulatory effect on autophagy in human colon cancer cells[118], but this stimulatory effect could be counteracted by GoLoco motifs. The Gαi3 protein and its partners (GAIP and AGS3) act prior to the formation of the autophagosome. They may be involved in the control of membrane flux to the autophagic pathway. What is noticeable is that the Gαi3 protein and its partner proteins can control the balance between the flow of membranes in the exocytic pathway and the delivery of membrane constituents to the macroautophagic pathway.
Monomeric G proteins Rab proteins are monomeric GTPases necessary for vesicular transport in the exo/endocytic pathway, in which Rab22a is associated with early and late endosomes. However, Rab22aQ64L, a mutant with low GTPase activity of Rab22a co-localizes with the autophagic vacuoles[69]. In addition, another monomeric GTPase, Rab24, existing preferentially in a GTP-bound state when expressed in cultured cells, is redistributed and co-localized with MAP1-LC3 during starvation. It has been shown that Rab24 has a role in autophagy. The GTP membrane-bound forms of Rab proteins are able to recruit cytosolic proteins, which target vesicles to appropriate sites on the acceptor membrane[119]. Whether or not this function of Rab proteins is essential for membrane fusion, transport, and the maturation of autophagic vesicles is still to be investigated.
Calcium Autophagy is dependent on the presence of sequestered Ca2+ in some intracellular storage compartments that are sensitive to interference by a number of Ca2+-perturbing agents[120]. ER is the major reservoir for intracellular calcium, thus thapsigargin, which inhibits the ER calcium/ATPase promotes the release of intracellular calcium from ER and thereby lowers ER calcium levels leading to notable inhibition of autophagy. This implies that depletion of sequestered, rather than of cytosolic, intracellular Ca2+ should be responsible for the common mechanism of autophagy inhibition. Inhibitors of Ca2+-activated protein kinases (KN-62, H-7, W-7) had little or no effect on autophagy, indicating that the Ca2+ requirement of autophagy was not mediated by these kinases. Recent studies have shown that elements that modify the lysosomal calcium levels, such as phorbol myristate acetate, ionophore A23187, and phentolamine, may modulate the total volume of autophagic vacuoles[121].
Protein synthesis pathway In rat hepatocytes, the phosphorylation of the ribosomal S6 protein, a p70S6 kinase substrate, is also related to the inhibition of autophagy. The degree of S6 phosphorylation determines the degree of occupancy of the endoplasmic reticulum by ribosomes, and thus determines the rate of autophagic sequestration and the rate of ER-linked protein synthesis[97]. So it is possible that there is a relationship between the control of protein synthesis and of autophagy. Furthermore, the finding of the involvement of eukaryotic initiation factor-2 alpha (eIF2α) kinases in autophagy strengthens this view. The eIF2a kinases are members of a family of evolutionarily conserved serine/threonine kinases that regulate stress-induced translational arrest. Recently, it was demonstrated that two eIF2α kinases (GCN2 and PKR) positively controled autophagic sequestration in yeast and mammalian cells in response to nutrient deprivation by phosphorylating the translation factor eIF2α[122]. However, a general inhibition of protein synthesis does not trigger autophagy. For example, when translation is inhibited by cycloheximide, autophagy is not induced, but the formation of smaller autophagosomes is observed. These results indicate that protein synthesis is required for the expansion of the preautophagosome vesicle[123]. In addition, the de novo synthesis of proteins is also required after the sequestration step during the maturation of autophagosomes.
Other regulatory factors The regulation of autophagy is very complex. In addition to the above mentioned molecular mechanisms, microtubule-associated protein, integrin, and some other kinases such as tyrosine protein kinase II, naringin-sensitive protein kinase, death-associated related protein kinase-1 (DRP-1), death-associated protein kinase (DAPK) all have a role[124]. However, the molecular mechanisms by which these regulatory factors contribute to the control of autophagy are still largely unknown and their downstream targets also remain to be identified.
Autophagy and diseases
Autophagy is an important gate-keeping mechanism for the stabilization of cell homeostasis, which is required for eliminating discarded or damaged organelles and/or cytoplasmic components and remodeling cytoplasm. Autophagy has been studied for more than 40 years, but it is only in the last 10 years that the molecular basis of autophagy has been gradually understood through the utilization of yeast genes. The discovery of the ATG genes and the dissection of the signaling pathways involved in the regulation of autophagy have greatly increased our knowledge of the occurrence and development of this lysosomal degradation pathway. In yeast, many questions about the molecular mechanisms of autophagy have already been investigated, but there are a great number of tasks ahead, such as clarification of the putative links between the different signaling complexes and elucidation of the specific mechanisms mediating autophagosome biogenesis, transport, fusion and autophagic degradation. Although the molecular machinery of the autophagic pathway is well conserved during evolution in multicellular organisms, especially in mammals, this process is much more complex in mammals than it is in yeast. At present, many autophagic genes in mammals still remain unknown and their functions in autophagy also remain undiscovered. The creation and analysis of transgenic and knockout animal models will help to understand the evolutionarily-acquired complexity of autophagy-mediated processes in mammals. Some gene (beclin-1, ATG5, and ATG7) knockout mice have already been used in experiments to explore the functions of these genes and the relationships between autophagy and some diseases[21,125,126]. For example, with a targeted beclin-1 mutant mouse model, it has been shown that heterozygous disruption of beclin-1 increases the frequency of spontaneous malignancies and accelerates the development of hepatitis B virus-induced premalignant lesions. In addition, it has also been demonstrated that beclin-1 is a haplo-insufficient tumor-suppressor gene, and therefore it is possible that the genetic disruption of autophagy, either by mutations of downstream autophagy-execution genes or by mutations in upstream autophagy-regulatory signaling pathways, may be an important mechanism of oncogenesis[126]. Thus, mutation of beclin-1 or other autophagy genes might contribute to the pathogenesis of human cancers. A growing number of pathological conditions, including cancer and neurodegenerative disorders, are associated with autophagy, so it is very important to reveal the molecular relationships between autophagy and diverse diseases. In our laboratory, we found that autophagy may have dual functions in cultured tumor cells. On the one hand, it may delay the apoptotic process in one cell type; but on the other hand, it may promote cell death in other cell types (Yan et al, unpublished observa-tions). Autophagy plays important roles in the degradation of misfolded or aggregated proteins and, therefore, may play a role in certain neurodegenerative diseases that feature the misfolding and aggregation of disease proteins, such as Huntington’s disease and Parkinson’s disease. We have studied the degradation of mutant huntingtin by autophagy. We found that autophagy upregulated cathepsins and enhanced the clearance of huntingtin fragments, but mutant huntingtin was relatively resistant to degradation by cathepsin D[127]. Over-stimulation of autophagy by mutant huntingtin resulted in mislocalization and dysfunction of mitochondria (Qin et al, unpublished observations). In our recent studies, we also found that an autophagic mechanism was involved in excitotoxicity. Activation of the NMDA (N-methyl-D-aspartate)- and KA (kainic acid)-type glutamate receptors stimulated autophagy and lysosomal enzymes. The apoptotic death of striatal neurons was blocked by 3-MA and a cathepsin B inhibitor, suggesting that the activation of autophagy probably contributes to excitotoxicity[128]. These studies have opened a new field for investigating the pathogenic mechanisms in neurodegenerative diseases related to protein misfolding, aggregation and excitotoxicity.
References
- Gronostajski RM, Pardee AB, Goldberg AL. The ATP dependence of the degradation of short- and long-lived proteins in growing fibroblasts. J Biol Chem 1985;260:3344-9.
- Scott SV, Hefner-Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky DJ. Cytoplasm-to-vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc Natl Acad Sci USA 1996;93:12304-8.
- Shieh HL, Chiang HL. In vitro reconstitution of glucose-induced targeting of fructose-1,6-bisphosphatase into the vacuole of semi-intact yeast cells. J Biol Chem 1998;273:3381-7.
- Lockshin RA, Zakeri Z. Apoptosis, autophagy and more. Int J Biochem Cell Biol 2004;36:2405-19.
- Bursch W, Ellinger A, Gerner C, Schulte-Hermann R. Autophago-cytosis and programmed cell death. In: Klionsky DJ, editor. Autophagy. Georgetown, TX: Landes Bioscience; 2004. p 287–303.
- Bergamini E, Cavallini G, Donati A, Gori Z. The anti-ageing effects of caloric restriction may involve stimulation of macro-autophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed Pharmacother 2003;57:203-8.
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C elegans. Science 2003;301:1387-91.
- Bergamini E, Cavallini G, Donati A, Gori Z. The role of macroautophagy in the ageing process, anti-ageing intervention and age-associated diseases. Int J Biochem Cell Biol 2004;36:2392-404.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004;306:990-5.
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 2002;27:421-9.
- Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003;5:539-45.
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun 2004;313:453-8.
- Wang CW, Klionsky DJ. Microautophagy. In: Klionsky DJ, editor. Autophagy. Georgetown, TX: Landes Bioscience; 2004. p 107–14.
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol 2004;36:2435-44.
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001;15:2286-7.
- Onodera J, Ohsumi YJ. Ald6p is a preferred target for autophagy in yeast, Saccharomyces cerevisiae. J Biol Chem 2004;279:16071-6.
- Dunn WA Jr. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol 1990;110:1923-33.
- Yamamoto A, Masaki R, Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J Histochem Cytochem 1990;38:573-80.
- Fengsrud M, Roos N, Berg T, Liou W, Slot JW, Seglen PO. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Exp Cell Res 1995;221:504-19.
- Kim J, Huang WP, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol 2001;152:51-64.
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001;152:657-67.
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 2001;20:5971-81.
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature 1998;395:395-8.
- Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem 2001;276:1701-6.
- Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 1999;18:5234-41.
- Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 1999;18:3888-96.
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003;116:1679-88.
- Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 2002;277:18619-25.
- George MD, Baba M, Scott SV, Mizushima N, Garrison BS, Ohsumi Y, et al. Apg5p functions in the sequestration step in the cytoplasm-to-vacuole targeting and macroautophagy pathways. Mol Biol Cell 2000;11:969-82.
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 1999;147:435-46.
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 2000;151:263-76.
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000;408:488-92.
- Ichimura Y, Imamura Y, Emoto K, Umeda M, Noda T, Ohsumi Y. In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J Biol Chem 2004;279:40584-92.
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19:5720-8.
- Kabeya Y, Mizushima N, Yamamoto A, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004;117:2805-12.
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004;15:1101-11.
- Sagiv Y, Legesse-Miller A, Porat A, Elazar Z. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J 2000;19:1494-504.
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature 1999;397:69-72.
- Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem 2003;278:3671-8.
- Per ES, Daniel JK. Approaching the molecular mechanism of autophagy. Traffic 2001;2:524-31.
- Scott SV, Nice DC 3rd, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem 2000;275:25840-9.
- Kim J, Kamada Y, Stromhaug PE, Guan J, Hefner-Gravink A, Baba M, et al. Cvt9/Gsa9 functions in sequestering selective cytosolic cargo destined for the vacuole. J Cell Biol 2001;153:381-96.
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000;150:1507-13.
- Uetz P, Giot L, Cagney G, Mansfield TA, Knight JR, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000;403:623-7.
- Nice DC, Sato TK, Stromhaug PE, Emr SD, Klionsky DJ. Cooperative binding of the cytoplasm to vacuole targeting pathway proteins, Cvt13 and Cvt20, to phosphatidylinositol 3-phosphate at the pre-autophagosomal structure is required for selective autophagy. J Biol Chem 2002;277:30198-207.
- Abeliovich H, Zhang C, Dunn WA Jr, Shokat KM, Klionsky DJ. Chemical genetic analysis of Apg1 reveals a non-kinase role in the induction of autophagy. Mol Biol Cell 2003;14:477-90.
- Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, et al. Interaction of the Unc-51-like kinase and microtubule associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res 2000;85:1-12.
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982;79:1889-92.
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000;275:992-8.
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997;243:240-6.
- Herman PK, Emr SD. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol Cell Biol 1990;10:6742-54.
- Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 2001;152:519-30.
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macro-autophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 2001;276:35243-6.
- Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoino-sitide 3-kinase-Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J 2003;376:577-86.
- Wurmser AE, Emr SD. Novel PtdIns(3)P-binding protein Etf1 functions as an effector of the Vps34 PtdIns 3-kinase in autophagy. J Cell Biol 2002;158:761-72.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999;402:672-6.
- Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ 2000;7:1263-9.
- Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, et al. Apg9/Cvt7 is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 2000;148:465-79.
- Kim J, Huang WP, Stromhaug PE, Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem 2002;277:763-73.
- Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 2000;148:465-80.
- Hagal A, Daniel JK. Autophagy in yeast: Mechanistic insights and physiological function. Microbiol Mol Biol Rev 2001;65:463-79.
- Shintani T, Suzuki K, Kamada Y, Noda T, Ohsumi Y. Apg2p functions in autophagosome formation on the perivacuolar structure. J Biol Chem 2001;276:30452-60.
- Guan J, Stromhaug PE, George MD, Habibzadegah-Tari P, Bevan A, Dunn WA Jr, et al. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol Biol Cell 2001;12:3821-38.
- Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 2004;15:231-6.
- Nice DC, Sato TK, Stromhaug PE, Emr SD, Klionsky DJ. Cooperative binding of the cytoplasm to vacuole targeting pathway proteins, Cvt13 and Cvt20, to phosphatidylinositol 3-phosphate at the preautophagosomal structure is required for selective autophagy. J Biol Chem 2002;277:30198-207.
- Stromhaug PE, Reggiori F, Guan J, Wang CW, Klionsky DJ. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell 2004;15:3553-66.
- Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem 2004;279:29889-94.
- Wang CW, Stromhaug PE, Kauffman EJ, Weisman LS, Klionsky DJ. Yeast homotypic vacuole fusion requires the Ccz1-Mon1 complex during the tethering/docking stage. J Cell Biol 2003;163:973-85.
- Mesa R, Salomon C, Roggero M, Stahl PD, Mayorga LS. Rab22a affects the morphology and function of the endocytic pathway. J Cell Sci 2001;114:4041-9.
- Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 2002;3:472-82.
- Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct Funct 2002;27:29-37.
- Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 2004;117:2687-97.
- Blankson H, Holen I, Seglen PO. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp Cell Res 1995;218:522-30.
- Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci 2000;113:1189-98.
- Nakamura N, Matsuura A, Wada Y, Ohsumi Y. Acidification of vacuoles is required for autophagic degradation in the yeast, Saccharomyces cerevisiae. J Biochem 1997;121:338-44.
- Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem 2001;276:2083-7.
- Suriapranata I, Epple UD, Bernreuther D, Bredschneider M, Sovarasteanu K, Thumm M. The breakdown of autophagic vesicles inside the vacuole depends on Aut4p. J Cell Sci 2000;113:4025-33.
- Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 2001;114:2491-9.
- Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem 2000;275:27447-56.
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in Lamp-2-deficient mice. Nature 2000;406:902-6.
- Cuervo AM, Mann L, Bonten EJ, d’Azzo A, Dice JF. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J 2003;22:47-59.
- Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 1999;13:3271-9.
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004;6:463-77.
- Jiang Y, Broach JR. Tor proteins and protein phosphatase 2A reciprocally regulate Tap42 in controlling cell growth in yeast. EMBO J 1999;18:2782-92.
- Rohde J, Heitman J, Cardenas ME. The TOR kinases link nutrient sensing to cell growth. J Biol Chem 2001;276:9583-6.
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 1999;402:689-92.
- Huang WP, Scott SV, Kim J, Klionsky DJ. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem 2000;275:5845-51.
- Chan TF, Bertram PG, Ai W, Zheng XF. Regulation of APG14 expression by the GATA-type transcription factor Gln3p. J Biol Chem 2001;276:6463-7.
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 2002;16:1472-87.
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004;7:167-78.
- Blommaart EF, Luiken JJ, Meijer AJ. Autophagic proteolysis: control and specificity. Histochem J 1997;29:365-85.
- Klionsky DJ. Regulated self-cannibalism. Nature 2004;431:31-2.
- Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003;1603:113-28.
- Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signaling by intracellular amino acid availability. Biochem J 2003;372:555-66.
- van Sluijters DA, Dubbelhuis PF, Blommaart EF, Meijer AJ. Amino-acid-dependent signal transduction. Biochem J 2000;351:545-50.
- Luiken JJ, Blommaart EF, Boon L, van Woerkom GM, Meijer AJ. Cell swelling and the control of autophagic proteolysis in hepatocytes: involvement of phosphorylation of ribosomal protein S6? Biochem Soc Trans 1994;22:508-11.
- Blommaart EF, Luiken JJ, Blommaart PJ, Vanwoerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995;270:2320-6.
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 1998;273:14484-94.
- Xu G, Marshall CA, Lin TA, Kwon G, Munivenkatappa RB, Hill JR, et al. Insulin mediates glucose-stimulated phosphorylation of PHAS-I by pancreatic beta cells. An insulin-receptor mechanism for autoregulation of protein synthesis by translation. J Biol Chem 1998;273:4485-91.
- McDaniel ML, Marshall CA, Pappan KL, Kwon G. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes 2002;51:2877-85.
- Meijer AJ, Dubbelhuis PF. Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun 2004;313:397-403.
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem 2003;278:16667-74.
- Plomp PJ, Gordon PB, Meijer AJ, Hoyvik H, Seglen PO. Energy dependence of different steps in the autophagic-lysosomal pathway. J Biol Chem 1989;264:6699-704.
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 1998;67:821-55.
- Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of AMP-activated protein kinase. J Biol Chem 1998;273:23758-63.
- Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells 2003;8:65-79.
- Wilson WA, Mahrenholz AM, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol 2001;21:5742-52.
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 2001;26:657-64.
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 2000;346:561-76.
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, et al. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 2002;4:699-704.
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003;11:1457-66.
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001;2:330-5.
- Liang XH, Yu J, Brown K, Levine B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. J Cancer Res 2002;61:3443-9.
- Ogier-Denis E, Couvineau A, Maoret JJ, Houri JJ, Bauvy C, De Stefanis D, et al. A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J Biol Chem 1995;270:13-6.
- Pattingre S, Petiot A, Codogno P. Analyses of G-alpha-interacting protein and activator of G-protein-signaling-3 functions in macroautophagy. Methods Enzymol 2004;390:17-31.
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of G alpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem 2000;275:39090-5.
- Bernard ML, Peterson YK, Chung P, Jourdan J, Lanier SM. Selective interaction of AGS3 with G-proteins and the influence of AGS3 on the activation state of G-proteins. J Biol Chem 2001;276:1585-93.
- Pattingre S, De Vries L, Bauvy C, Chantret I, Cluzeaud F, Ogier-Denis E, et al. The G-protein regulator AGS3 controls an early event during macroautophagy in human intestinal HT-29 cells. J Biol Chem 2003;278:20995-1002.
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001;2:107-17.
- Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993;268:26107-12.
- Kalamidas SA, Kotoulas OB, Hann AC. Studies on glycogen autophagy: effects of phorbol myristate acetate, ionophore A23187, or hentolamine. Microsc Res Tech 2002;57:507-11.
- Tallóczy Z, Jiang W, Virgin-IV HW, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad Sci USA 2002;99:190-5.
- Abeliovich H, Dunn WA Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 2000;151:1025-34.
- Petiot A, Pattingre S, Arico S, Meley D, Codogno P. Diversity of signaling control of macroautophagy in mammalian cells. Cell Struct Funct 2002;27:431-41.
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003;112:1809-20.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005;169:425-34.
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, et al. Autophagy regulates processing amino terminus huntingtin fragments. Hum Mol Gen 2003;12:3231-44.
- Wang Y, Gu ZL, Cao Y, Liang ZQ, Rong H, Bennett MC, et al. Lysosomal enzyme cathepsin B is involved in kainic acid-induced excitotoxicity in rat striatum. Brain Res 2005;1039:203-6.