
Molecular mechanisms regulating expression and function of transcription regulator “inhibitor of differentiation 3”
Introduction
Inhibitor of differentiation (Id) proteins are a family of transcriptional regulators that have been implicated in several developmental, physiological and pathological processes. Their name relates to their ability to inhibit the differentiation of a variety of cells by inhibiting the DNA binding activity of many transcription factors that regulate expression of cell-type specific genes. Id genes are widely expressed in the animal kingdom from humans to zebra fish[1]. Four Id genes, Id1–Id4 have been found in humans and in rodents. A homologous Id-like gene, extramacro-chaetae has been identified in drosophila[2].
The Id proteins are small proteins of approximately 13 kDa–20 kDa. All 4 Id proteins contain a relatively conserved helix-loop-helix (HLH) structural motif in the middle of the protein, but are otherwise quite divergent in sequences. The 4 Id proteins constitute 1 subclass (Class V) of the large family of HLH transcriptional regulators. Unlike other HLH proteins that can bind to DNA as either homodimers or heterodimers, the Id proteins lack the basic amino acid domain needed for DNA binding. Instead, they are believed to function primarily by forming heterodimers with the “ubiquitous” Class I HLH proteins known as E-proteins. This prevents the E-proteins from interacting with each other and with the cell-type specific Class II HLH proteins, inhibiting their binding to DNA and blocks their ability to modulate gene expression (Figure 1).
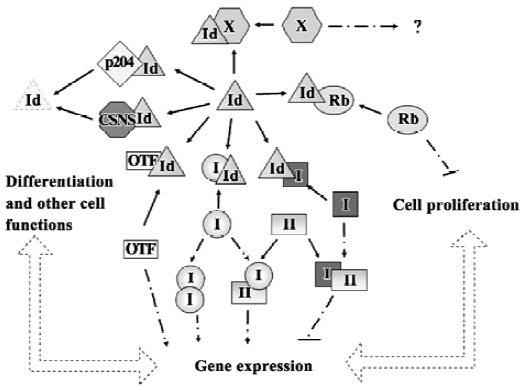
The first Id gene, Id1, was identified by virtue of the ability of its encoded protein to inhibit muscle differentiation and the activation of muscle-specific gene promoters[3]. However, the Id proteins are now known to be important in other physiological systems and pathophysiological situa-tions. In addition to the E-protein partners, Id proteins regulate other transcription factors such as ternary complex factor (TCF)/ETS[4,5], Pax 5 and sterol regulatory element binding protein-1[6]. Individual Id proteins might also interact selectively with proteins not recognized by other Id family members. For example, Id1 is the only Id protein shown to bind the proteasomal protein S5a[7]. Similarly, only Id2 binds to the tumor suppressor retinoblastoma protein Rb and interferes with the ability of hypophosphorylated Rb to suppress cell proliferation when both are ectopically expressed (Figure 1)[8].
Evidence to date indicates that the Id proteins are likely to carry out both common and distinct biological functions, depending in part on when and where the proteins are expressed. Consistent with this idea, mice engineered to be deficient in a single Id gene are viable, albeit with developmental defects in certain specific cell lineages depending on the particular gene that has been inactivated[9–15]. For example, Id3-deficient mice have defects in both B cell[14] and T cell[15] maturation and develop salivary gland defects reminiscent of the autoimmune Sjogren’s syndrome, but are otherwise generally normal[16]. In contrast, mice with both copies of Id1 and Id3 genes inactivated die in utero (embryonic d 13.5) with severe vascular defects in the forebrain, aberrant neuronal differentiation[17] and multiple cardiac abnormalities[18]. In fact, mice with both copies of any 2 of the 3 Id genes (Id1–Id3) inactivated exhibit similar cardiac defect and are embryonic lethal. Mice with loss of 1–3 copies of the Id1/1d3 genes are viable but exhibit increasing degrees of resistance to tumor-induced angiogenesis[17]. The lethality of the double knockout mice and the dosage-dependent tumor angiogenesis phenotype clearly indicates that the Id genes mediate overlapping function in some developmental lineage. However, the developmental defects in specific cell lineages manifested in single knockouts suggest that Id genes are unable to compensate in some cells, perhaps because they are not co-expressed or, as in the case of Id2, because of specific dimerization with different protein partners such as Rb.
Many excellent and extensive reviews covering the Id family proteins have been published in recent years[19–26]. Most of these reviews have dealt with the Id proteins as a group and concentrated primarily on the potential biological functions of the Id proteins. Relatively less attention has been devoted to reviewing the molecular mechanisms that regulate the expression and function of individual Id genes and proteins.
The present review will focus on the third member of the Id gene family, Id3, particularly on the mechanisms involved in its regulation. The Id3 gene was first identified as a serum-inducible immediate early gene in an established murine fibroblastic cell line[27]. Subsequent studies have documented its involvement in various biological processes, including T and B cell development[15,14], skeletal muscle differentia-tion[28,29], vascular smooth muscle cell proliferation[30,31], embryonic neurogenesis[17], osteogenesis[32] and tumor-induced angiogenesis[17].
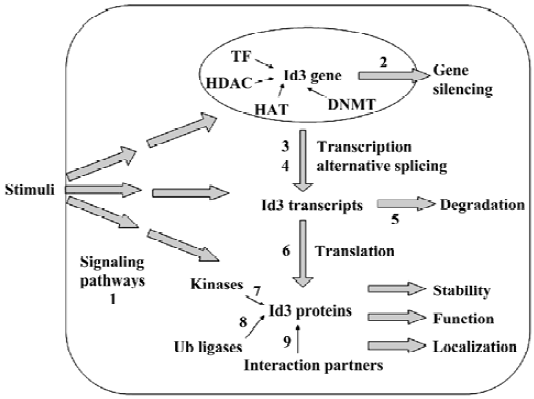
Expression and function of the protein is under many complex layers of regulation (Figure 2) and, therefore, could provide rich targets for therapeutic interventions.
Developmental and cell-type specific expres-sion pattern of Id3
The most direct way to regulate the function of a particular protein is to control when and where the gene encoding the particular protein is expressed. Several studies have characterized the expression of Id3 at either the mRNA or the protein level. A wide range of techniques have been utilized, including Northern, in situ hybridization, reverse transcription with polymerase chain reaction, various genome expression profiling assays, Western immunoblots and immunocytochemical staining procedures.
Like other Id genes, the expression of Id3 is dynamically regulated during embryonic development. The general expression level is high at the early embryonic ages, but progressively declines as the embryo develops[27,33]. Id3 is widely expressed throughout the embryo proper. Its expression is readily detectable within regions that are undergoing active morphogenesis[34], but can also be detected in some undifferentiated tissues[33].
The expression pattern of Id3 during embryonic development overlaps with, but is not identical to that of, the other Id genes. At an early embryonic age, Id1 and Id3 are expressed in the tissues derived from the inner cell mass; Id2 is expressed in tissues derived from trophoblasts; but no Id4 expression is evident. In the primitive gut, Id1 and Id3 signals are expressed in the mesenchyme, whereas Id2 expression occurs within the epithelium[34]. During early spinal cord development, expression of Id1 and Id2 are restricted to the roof plate, whereas Id3 is expressed both in the roof and the floor plate. At later stages, Id1 and Id3 expression is detected in the dividing neuroblasts, whereas Id2 and 4 are expressed in presumptive neurons undergoing matura-tion[35]. Generally speaking, the pattern of Id3 expression most closely resembles that of Id1, but the coincidence is not absolute[33].
Expression of Id3 in adult tissues is widely spread but not universal and the level of expression varies substantially among different tissues. Id3 is expressed in many cell types in vivo and in both primary cultures and established cell lines (Table 1). Depending on the cell and tissue type, the expression might be constitutive or detectable only after exposure to appropriate stimulus. The level of Id3 expression is generally high in proliferating, undifferentiated cells, but downregulated when cells undergo terminal differentia-tion[28,29,36–39]. Expression of Id3 also tends to be higher in immortalized cell lines, consistent with the putative involvement of Id proteins in combating cellular senescence[40,41] and maintaining the capacity for self-renewal in embryonic stem cells[42].
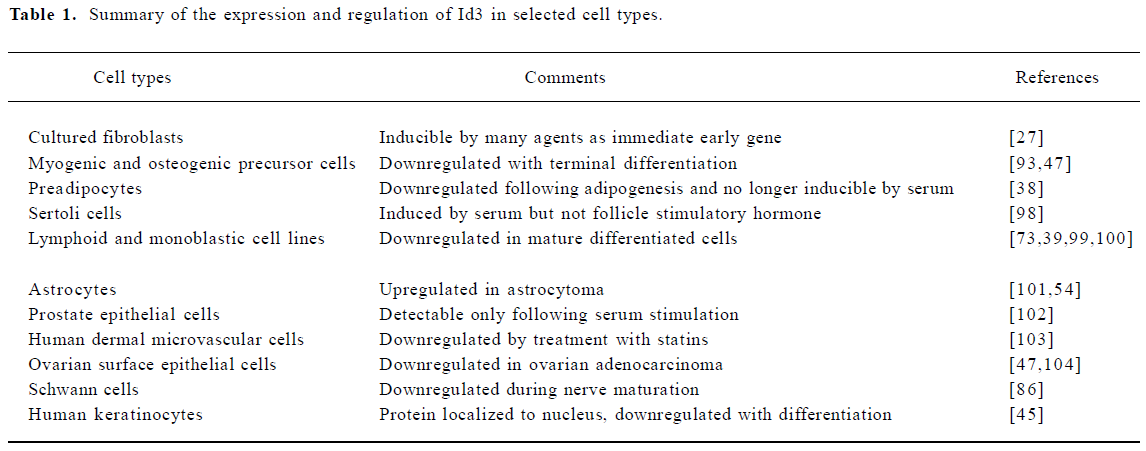
Full table
Knowing what tissues and cell types express Id3 is, however, probably akin to just seeing the tip of the iceberg. The level of Id3 expression is not static, but varies dramatically with the growth and physiological state of the cells and is modulated in response to diverse extracellular stimuli. There are other layers of regulation superimposed upon the regulation at the expression level (Figure 2). Additional caveats should also be kept in mind when interpreting the results from the aforementioned studies. Results using immortalized cell lines need to be viewed with caution because the process of immortalization might involve alterations in Id3 expression not seen in vivo. Direct examinations of tissue sections in vivo or with primary cell cultures eliminate such concerns. However, the sensitivity of the assays used for detecting Id3 expression in various studies are not precisely known and might not always be comparable. The specificity of the antibodies used for immunodetection might not have always been adequately scrutinized. Therefore, care should be taken in making a firm conclusion as to whether Id3 is or is not expressed in a certain tissue or cell type. Nevertheless, because Id3 is expressed at many, but not all, cells indicates that its regulation is likely to involve both ubiquitous as well as cell-type specific regulatory mechanisms.
Altered patterns of Id3 expression in diseases and pathophysiological situations
Perturbation of Id3 expression has been correlated with a variety of disease states and pathological situations, including cancer, aging, atherosclerosis, muscle atrophy, and inflammation. Conversely, altered expression of Id3 has been detected during the regenerative process following tissue injury.
It is generally believed that members of the Id gene family behave like oncogenes. Overexpression of one or more Id genes has been detected in various cancers. The situation with Id3 is consistent in most part with this generalization[43–45], but there are some exceptions. In certain neurological tumors, Id3 upregulation is observed not only in the tumors themselves but also in the vascular tissues surrounding the tumors[44]. In contrast, expression is reduced in papillary thyroid carcinoma[46] and ovarian carcinomas[47], and either increased[48] or absent[49] in seminoma. The expression pattern is even more complex during the development of liver diseases and liver cancer. Id3 expression is low in normal liver, increases with the progression of liver diseases from chronic hepatitis to liver cirrhosis and is expressed at high levels in well-differentiated hepatocarcinomas, but not in the more advanced de-differentiated tumors[50].
Variations in Id gene expression have also been correlated with aging in animals. Expression of Id1, Id2, and Id3 increases in hind limb muscles of aging rats[51]. Because increased levels of Id proteins are associated with muscle disuse atrophy[52], the upregulated Id expression in aging muscle might contribute to the loss of muscle mass that commonly accompanies aging. In contrast, Id3 expression appears to be reduced in the pituitary gland in aged rats[53]. Taken together, the results suggest that some other aging-related physiological perturbations rather than aging per se might be responsible for the altered Id expression in different tissues.
Id3 expression level also changes in inflammatory and atherogenic processes. Id gene expression is upregulated in reactive astrocytes activated as part of the inflammatory process following spinal cord injury[54]. Id3 expression is also altered in vascular smooth muscle cells (VSMC) during atherogenesis. It is expressed at low level in normal vessels of the carotid artery, but is increased within 3 d of balloon injury and remains high through 14 d postinjury[30]. This is accompanied by the appearance of a novel differentially spliced Id3 transcript.
Changes in Id3 expression are not limited to pathological situations: increased Id3 expression has been implicated in tissue regeneration. In the African clawed frog, Xenopus laevis, which does not completely regenerate the missing limb following amputation, Id3 expression is upregulated transiently but returns to basal level when terminal differentiation of the limb stump tissues is initiated. In the Japanese newt, Cynops pyrrhogaster, which is capable of regenera-ting a complete limb, expression of Id3 persists until the stage of digit formation[55]. The earlier downregulation of Id3 in the Xenopus might have contributed to premature differentiation and the aborted regeneration program. Similarly, Id3 upregulation might also contribute to the liver regeneration following partial hepatomy in mice[56].
The mechanisms accounting for the perturbation of expression of Id3 under different conditions are largely unknown. The presumption is that it involves changes in transcription, but the hypothesis is as yet unproven. No evidence has been found so far that would implicate either gene amplification or deletion in the altered pattern of Id3 expression found in different cancers; therefore, the change is probably epigenetic. There is some evidence that DNA methylation or histone acetylation might contribute to Id3 regulation but exactly how such processes affect Id3 expression is not known (see below). In most cases, the change in Id3 expression is likely to be either secondary to changes in the cell physiology and/or in response to environmental signals.
Stimulus and signaling pathways regulating Id3 expression
Id3 was originally identified as a serum-inducible gene in Balb/c3T3 fibroblasts and found to be induced in these cells, to a varying degree, by a variety of other agents[27]. The Id3 transcript was induced rapidly and transiently, with peak accumulation occurring at about 1 h after stimulation. In this system, Id3 appears to behave as “immediate-early” or “primary response” genes, and can be induced in the absence of ongoing protein synthesis.
Id3 expression has been shown to be responsive to even more diverse stimuli in a variety of cell types (Table 2). Several conclusions can be drawn from this plethora of information. First, although the ability to regulate Id3 expression is wide spread, not all agents regulate Id3 expression in the same manner in all cell types. For example, the phorbol ester, phorbol 12-myristate 13-acetate (PMA), stimulates Id3 expression in thyrocytes[46] but downregulates Id3 levels in the SH-SY5Y neuroblastoma cells[57]. This suggests that the regulatory pathways might be different among different cell types. Second, the temporal pattern of Id3 expression is very complex, and differs depending on the cell type and stimuli. In many cases, the induction is transient but biphasic, with an early peak of expression followed either by a later second peak or conversely by long-term suppression. In other cases, the change appears to be more persistent. For example, Id3 is induced transiently by transforming growth factor-β (TGF-β) in B-lymphocyte progenitors, but returns to basal level within 20 h[13]; whereas in epithelial cells, the transient increase is followed by long-term suppression[58,59]. Id3 is also transiently induced by bone morphogenetic proteins (BMP) in mesenchymal stem cells[60], but a sustained induction is observed following BMP treatment of epithelial cells[59]. Finally, and perhaps most importantly, the differences in the patterns of expression have immense biological significance. For example, insulin, which is an adipogenic agent, induces Id3 transiently in 3T3L1 preadipocytes, but adipogenic differentiation itself is accompanied by long-term downregulation of Id3 in 3T3F24A cells[38]. Similarly, B cell receptor (BCR) engagement in mature B cells results in transient Id3 induction and stimulation of cell proliferation[14]. PMA, which is also mitogenic, results in a biphasic response with a transient peak at 4 h and a secondary peak at 24 h, around the time when the stimulated cells are entering S[61]. In contrast, a more persistent Id3 expression is induced by BCR engagement in immature B cells and results in the inhibition of cell proliferation[62].
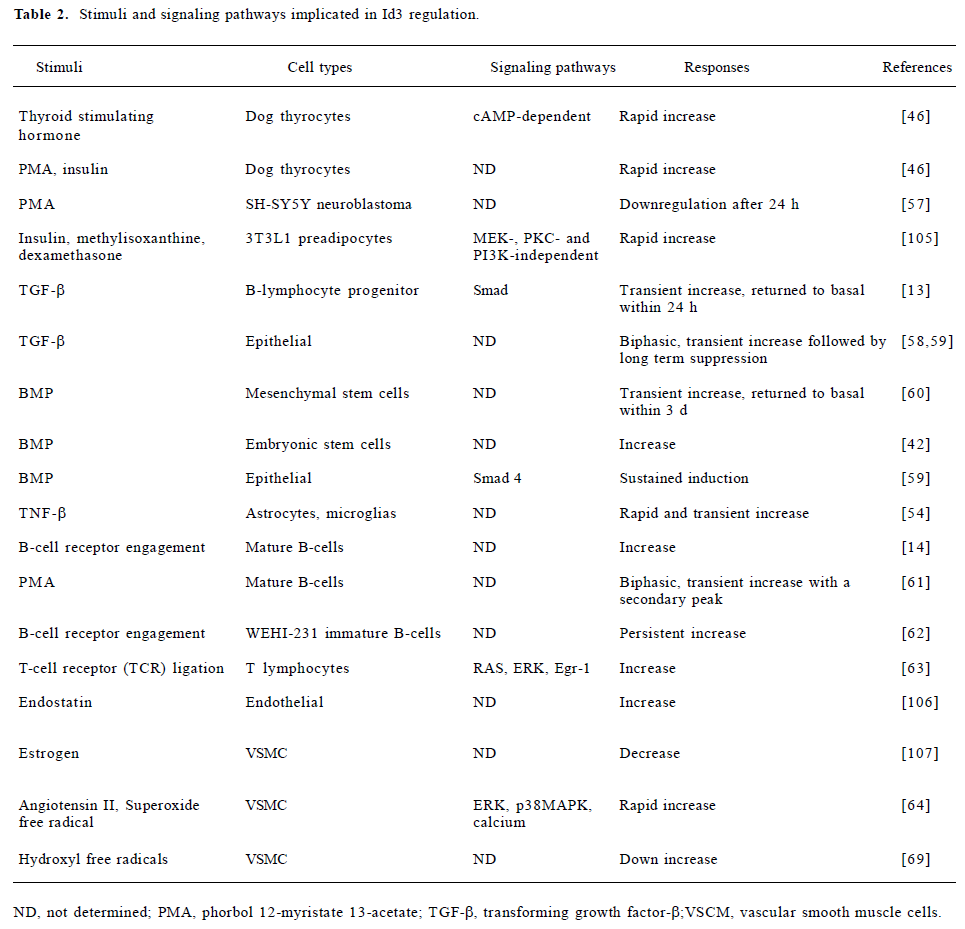
Full table
Because Id3 expression is regulated by many cytokines, hormones and other environmental signals, several different signal transduction pathways presumably contribute to its regulation. Pathways that have been implicated include the Ras, the ERK1/2 pathway following TCR engagement[63], the Smad-dependent pathway in response to TGF-β and BMP[59,13] and the p38, ERK and calcium-dependent pathways following superoxide free radical-induced oxidative stress[64]. In the case of TGF-β, the biphasic response could reflect the differential actions of 2 classes of TGF-β receptors (Activin receptor-like kinase 1 and Activin receptor-like kinase 2) that are preferentially coupled to different Smad proteins[65]. In most cases, the precise signaling pathway(s) responsible for regulating Id3 expression has not been completely defined. In addition, it has not been conclusively demonstrated that the change in transcript level reflects authentic transcriptional regulation.
Regulatory elements and transcription factors involved in Id3 promoter regulation
A systematic analysis of the Id3 promoter and the mechanisms involved in its regulation has yet to be carried out. Yeh and Lim reported in 2000 the cloning and initial characterization of the promoter region of the mouse Id3 gene extending approximately 1 kb upstream of the transcription start site[66] and found that a 180 bp proximal Id3 promoter fragment was sufficient for substantial transcriptional activity in the proliferating myogenic cell line C2C12[66]. Recently, a 254 bp Id3 promoter fragment (-200/+54) has been shown also to be transcriptionally active and responsive to BCR engagement in the WEHI-231 immature lymphoid cells[67]. Several putative transcription factor binding sites have been identified by computer-based analysis, but whether they are bona fide regulatory motifs has not been determined (Figure 3). Using in vitro DNase protection assay, we have identified 2–3 protected footprints within this region, at least 1 of which (site 1) seems to be responsible for much of the Id3 promoter activity seen in proliferating C2C12 cells[67a]. Moreover, electrophoretic mobility shift assays detected proteins in the nuclear extracts of proliferating C2C12 cells that bind to the 180 bp Id3 promoter frag-ment[66]. A mutation that eliminates the DNA binding to site 1 reduced the transcriptional activity of the Id3 promoter, indicating that the site is likely to be functional. Consistent with this idea, protein binding to the site declined substantially when incubated in vitro with nuclear extracts isolated from differentiated muscle cells that no longer express Id3[68]. It is yet to be determined whether the site in the endogenous Id3 promoter is occupied by transcription factor in vivo.
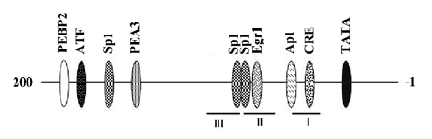
The second DNase footprint (site 2) encompasses a previously identified early growth response factor 1 (egr-1) site that has been shown to bind recombinant egr-1 in vitro[27]; whether the site binds egr-1 in vivo has not been established. Others have reported that the upregulation of Id3 upon TCR engagement involves egr-1, but direct egr-1 binding to the promoter has not been demonstrated and the egr-1 binding site responsible for the upregulation of Id3 promoter activity has not been localized[63]. We have found, however, that mutating the egr-1 site in footprint 2 does not affect the promoter activity of the proximal promoter in proliferating C2C12 cell[68]. Either other sites are involved in mediating egr-1 dependent activation of the Id3 promoter or the mechanism of promoter regulation differs between the muscle and T-cell systems.
Several other transcription factors have been reported to either directly or indirectly regulate Id3 expression. The zinc finger transcription factor, gut-enriched kruppel-like factor (GKLF), downregulates the expression of Id3 in response to hydroxyl free radicals in VSMC, apparently by binding to a GKLF site in the Id3 promoter[69]. Likewise, the transcriptional repressor, B lymphocyte induced maturation protein-1, also appears to act directly on the Id3 promoter region as demonstrated by the chromatin immunoprecipitation assay in mature B-lymphoblast cells[70].
Exactly how the other transcription factors affect Id3 expression is not known. For example, ectopic expression of myoD in proliferating muscle cells upregulates Id3[71], but it is not known whether this involves direct binding of myoD to the Id3 promoter. We did not find any E-box motif corresponding to myoD binding sites within the 1 kb region that we have analyzed, but interactions at more distal sites have not been ruled out. Other transcription factors, such as the homeodomain protein AT binding factor 1A (ATBF1A), the lymphocyte specific transcription coactivator BOB.1/OBF.1, and the forkhead transcription factor FOXM1B, have all been shown to either up or downregulate Id3 expression when overexpressed, but in no case has direct interaction between these factors and the Id3 promoter been establish-ed[72,73,56]. Cells that are deficient in Smad4 fail to alter their Id3 level in response to either TGF-β or BMP, suggesting that the Smad proteins are involved in mediating the regulatory effect of these cytokines[59,60]. However, direct binding of Smad on the Id3 promoter has not been demonstrated.
Several lines of evidence suggest that DNA methylation and histone acetylation might either directly or indirectly regulate Id3 expression. In some non-lymphoid hematopoietic cell lines, the lack of Id3 expression has been correlated with hypermethylation of DNA upstream of the Id3 transcription start site[74]. Whether similar methylation events take place in other cases where Id3 expression is silenced is not known. Treatment with 5 azacytidine, an inhibitor of DNA methyl transferase, blocks the neuronal differentiation of PC12 cells induced by NGF and inhibits NGF-induced Id3 downregulation that occurs during differentiation[36]. Conversely, Id3 expression is downregulated in lymphoblastic cells from patients with immunodeficiency, centromere instability and facial anomalies syndrome, a disease syndrome caused by a defective DNA methyltransferase (DNMT3B)[75]. It is not yet known whether the methylation state of the Id3 promoter itself is directly affected in either case. Similarly, addition of histone deacetylase inhibitors to lung adnocarcinoma cells and the K562 hematoprogenitor cells upregulates Id3 expression, but whether this represents a direct effect on the histones associated with the Id3 promoter is not clear[76–78].
Regulation of Id3 isoform expression by differential splicing
The Id3 gene is composed of three exons and two introns. The first exon includes the 5' untranslated region (UTR) and the coding region corresponding to the first 100 amino acid residues. The coding region is interrupted by a 105 bp intron at the +357 position of the mouse Id3 gene. The second exon contains the last 19 amino acid coding region and 26 bp of the 3' UTR of the mouse Id3 transcript. In the mouse gene, this is followed by a 506 bp second intron and the third exon coding for the rest of the UTR[66]. Both the human and rat Id3 genes appear to be similarly organized, although the length and exact nucleotide sequence of the introns vary substantially among species.
It has been shown in both rats and human that an alternative transcript can be generated under certain conditions by the retention of the first intron[79,31], resulting in the production of a novel Id3 protein isoform with an altered C-terminus. However, because of the divergent nucleotide sequences in the intron, the novel Id3 C-terminal peptides vary substantially in both length and sequences across species. In humans, the novel terminus is 60 amino acids in length; whereas in mice and rats it is only 29 amino acids long, with 7 out of the 29 amino acids differing between the two species. In contrast, the original protein isoform from the mouse and rat differs by only 1 amino acid substitution out of 119.
With such divergence in sequence among closely related species, one might have predicted that the novel isoforms (referred to as Id3L in humans and Id3a in rats) would have dubious biological significance. Contrary to this expectation, in situ hybridization and immunolocalization using isoform-specific antibody indicates that the novel variant is not produced in normal vasculature, but is dramatically upregulated during the later stages of vascular lesion formation, whereas the original Id3 variant is upregulated earlier in the neoin-tima[30]. Furthermore, the 2 Id3 protein variants appear to be functionally distinct. The human Id3L protein is a weaker inhibitor of the binding of E-proteins to DNA in vitro than the original shorter Id3 isoform[31]. Overexpression of the original shorter rat Id3 protein in VSMC promotes proliferation and S-phase entry and inhibits transcription of the cyclin dependent kinase (cdk) inhibitor p21Cip1, whereas overproduction of the novel Id3a does not inhibit p21 transcription, and causes a decrease in cell number, presumably by promoting apoptosis[30].
What accounts for the unique biological functions of the novel Id3 isoform is perplexing, because the C-termini encoded by the non-spliced Id3 variants would be very different between humans and rats and differ substantially even between the more closely related rodent species. One possible explanation of this conundrum is that the altered C-termini might lead to a protein that acts essentially like a “dominant negative” mutant. In support of this possibility, we have shown that truncation of the Id3 C-terminal to a site roughly corresponding to the spliced junction resulted in a protein that was incapable of blocking E-protein binding, and the inhibition of E-box dependent transcription was compromised[80]. Swapping the Id3 C-terminal with the corresponding region of the Id2 C-terminal likewise reduced the ability of the fusion protein to block E-protein activity. Why the C-terminus of the Id3 protein is so critical for its activity is presently unknown. Much remains to be learned about how the alternative splicing is regulated and when and where else the novel Id3 variant might be expressed. Because most of the earlier studies did not take into account the existence of the novel isoform, some of the expression data might need to be revisited.
Regulation of Id3 transcript and protein stability
Like many other immediate early genes, the Id3 transcript has a rather short half-life[81]. Examination of the cDNA sequence revealed the presence of instability elements in the 3' UTR that bind to RNA-binding protein[82,83]; sequence elements that regulate cytoplasmic polyadenylation are also present[84]. These sequence motifs are likely to contribute to the regulation of the stability and/or translation of the Id3 transcripts, but detailed studies have yet to be reported.
The Id3 protein is also rather short lived, with a half-life of approximately 20 min when overexpressed in 293 cells. As with other short-lived proteins, Id3 appears to be degraded by proteasomes in an ubiquitination dependent manner[85]. Treatment with proteasome inhibitors stabilizes Id3, allowing it to accumulate. Cells with mutations in the E1 ubiquitin activating enzyme show increased Id3 stability. Co-expression of the bHLH protein E47 with Id3 significantly reduces the rate of degradation of Id3[86]. In contrast, interaction with the interferon-inducible protein p204, which is upregulated during terminal muscle differentiation, enhances Id3 degradation[87], suggesting that the stability of the Id3 protein might be affected by its dimerization partner.
Whether and how ubiquitination of Id3 is regulated by the cellular environment is not known. Id3 has been shown to interact physically with CSN5, a subunit of the COP9 signalsome complex that has been implicated in the regulation of the ubiquitination of a large number of cellular proteins. Inhibitors of the COP9-associated kinases increase the ubiquitination of Id3 and accelerate its degradation, whereas overexpression of another COP9 subunit CSN2 stabilizes the Id3 protein. Given these data, it has been speculated that CSN-mediated phosphorylation inhibits ubiquitination of Id3[88].
Although the role of ubiquitination in Id3 degradation is now well-documented, we found that replacing all 4 lysine found in the mouse Id3 protein with arginine failed to stabilize the protein. This suggests that Id3 might be added to an increasing list of proteins like myoD[89], Id2[90], and p21cip1[91], that gets ubiquitinated at the α-amino group of the N-terminal residue instead of an internal lysine residue.
How much the regulation at the level of protein stability contributes to the overall regulation of Id3 expression level is not presently known. In one study, experimental denervation to the muscle of young rats results in an increase in Id3 protein levels in both the soleus (900%) and gastrocnemius (80%) without apparently affecting the mRNA level[51]. Similar regulation might occur under other conditions.
Regulation of protein localization and function
Although Id3 preferentially interacts with the class I HLH proteins (E proteins) and exhibits little or no affinity towards the class II bHLH proteins[92], it also binds to other transcription factors such as TCF[4] and the homeodomain protein Pax5[6] through the HLH domain. Co-expression of E-proteins competes with Pax5 for binding to Id3. Other protein partners might also compete with each other for binding. The relative abundance of the various protein components could, therefore, affect the precise compositions of the protein complexes that are formed. This might in turn determine the biological effects of Id3 under conditions where Id3 level is limiting. In addition, because some E-protein isoforms can act as transcriptional inhibitors[93], Id3 might even be expected to activate gene expression under certain circums-tances.
Dimerization between Id3 and its target proteins might also affect its subcellular localization. When expressed alone in COS cells, Id3 is predominantly cytoplasmic/perinuclear. Co-expression with E-proteins causes it to accumulate in the nucleus[94]. Whether other proteins or mechanisms might control the subcellular distribution of Id3 is presently unknown. Interestingly, the Id3 protein has been reported to be localized to specific subcellular locations in some cell types[48,95], but the mechanism responsible remains unknown.
The Id3 protein contains several potential phosphorylation sites for various protein kinases[96], including a conserved putative cdk2 site around Ser5 in the N-terminus, which is phosphorylated in a cell cycle-dependent manner (during late G1 to S). Id3 proteins with a mutation of Ser5 to Asp5 to mimic phosphorylation are better able to block the formation of heterodimers between E-protein and myoD but are less able to block E-protein homodimerization. Phosphorylation by cdk2 also interferes with the ability of Id3 to bind to TCF proteins[5], suggesting that Ser5 phosphorylation might alter the dimerization specificity of Id3. In fibroblasts, the Id3 Asp5 mutant (mimicking phosphorylation) is unable to promote S-phase transition, whereas the non-phosphoryla-table Ala5 mutant is more effective than the wild type[61]. In contrast, in VSMC, the non-phosphorylatable Id3 Ala 5 mutant activates the cdk inhibitor p21cip1 promoter and is unable to promote an increase in cell number when over-expressed[97]. Why the behavior is different between the two cell types is unknown.
Conclusions
It is clear from this brief review that the expression and function of the Id3 protein are regulated in a very complex manner. Much remains to be learned about how the Id3 gene is regulated transcriptionally. Almost nothing is known about how the alternative splicing event that generates a potentially dominant negative protein is controlled. Posttranslational modifications might impact the stability, dimerization specificity and/or subcellular localization of the protein, which might then affect the function of the protein. In view of the many physiological and pathophysiological situations where Id protein function is involved or perturbed, understanding these issues will, no doubt, provide a fertile ground for the development of potential therapeutic interventions.
References
- Dickmeis T, Rastegar S, Lam CS, Aanstad P, Clark M, Fischer N, et al. Expression of the helix-loop-helix gene Id3 in the zebrafish embryo. Mech Dev 2002;113:99-102.
- Campuzano S. Emc, a negative HLH regulator with multiple functions in Drosophila development. Oncogene 2001;20:8299-307.
- Benezra R, Davis RL, Lassar A, Tapscott S, Thayer M, Lockshon D, et al. Id: a negative regulator of helix-loop-helix DNA binding proteins. Control of terminal myogenic differentiation. Ann NY Acad Sci 1990;599:1-11.
- Yates PR, Atherton GT, Deed RW, Norton JD, Sharrocks AD. Id helix-loop-helix proteins inhibit nucleoprotein complex formation by the TCF ETS-domain transcription factors. EMBO J 1999;18:968-76.
- Stinson J, Inoue T, Yates P, Clancy A, Norton JD, Sharrocks AD. Regulation of TCF ETS-domain transcription factors by helix-loop-helix motifs. Nucleic Acids Res 2003;31:4717-28.
- Roberts EC, Deed RW, Inoue T, Norton JD, Sharrocks AD. Id helix-loop-helix proteins antagonize pax transcription factor activity by inhibiting DNA binding. Mol Cell Biol 2001;21:524-33.
- Anand G, Yin X, Shahidi AK, Grove L, Prochownik EV. Novel regulation of the helix-loop-helix protein Id1 by S5a, a subunit of the 26 S proteasome. J Biol Chem 1997;272:19140-51.
- Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol 1996;16:2570-8.
- Volpert OV, Pili R, Sikder HA, Nelius T, Zaichuk T, Morris C, et al. Id1 regulates angiogenesis through transcriptional repression of thrombospondin-1. Cancer Cell 2002;2:473-83.
- Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol Cell Biol 1997;17:7317-27.
- Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 1999;397:702-6.
- Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 2003;4:380-6.
- Kee BL, Rivera RR, Murre C. Id3 inhibits B lymphocyte progenitor growth and survival in response to TGF-beta. Nat Immunol 2001;2:242-7.
- Pan L, Sato S, Frederick JP, Sun XH, Zhuang Y. Impaired immune responses and B-cell proliferation in mice lacking the Id3 gene. Mol Cell Biol 1999;19:5969-80.
- Rivera RR, Johns CP, Quan J, Johnson RS, Murre C. Thymocyte selection is regulated by the helix-loop-helix inhibitor protein, Id3. Immunity 2000;12:17-26.
- Li H, Dai M, Zhuang Y. A. T cell intrinsic role of Id3 in a mouse model for primary Sjogren’s syndrome. Immunity 2004;21:551-60.
- Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 1999;401:670-7.
- Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, et al. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science 2004;306:247-52.
- Sikder HA, Devlin MK, Dunlap S, Ryu B, Alani RM. Id proteins in cell growth and tumorigenesis. Cancer Cell 2003;3:525-30.
- Benezra R, Rafii S, Lyden D. The Id proteins and angiogenesis. Oncogene 2001;20:8334-41.
- Lasorella A, Uo T, Iavarone A. Id proteins at the cross-road of development and cancer. Oncogene 2001;20:8326-33.
- Zebedee Z, Hara E. Id proteins in cell cycle control and cellular senescence. Oncogene 2001;20:8317-25.
- Yokota Y, Mori S. Role of Id family proteins in growth control. J Cell Physiol 2002;190:21-8.
- Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol 2003;13:410-8.
- Alani RM, Silverthorn CF, Orosz K. Tumor angiogenesis in mice and men. Cancer Biol Ther 2004;3:498-500.
- Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci 2000;113:3897-905.
- Christy BA, Sanders LK, Lau LF, Copeland NG, Jenkins NA, Nathans D. An Id-related helix-loop-helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci USA 1991;88:1815-9.
- Atherton GT, Travers H, Deed R, Norton JD. Regulation of cell differentiation in C2C12 myoblasts by the Id3 helix-loop-helix protein. Cell Growth Differ 1996;7:1059-66.
- Melnikova IN, Christy BA. Muscle cell differentiation is inhibited by the helix-loop-helix protein Id3. Cell Growth Differ 1996;7:1067-79.
- Forrest ST, Barringhaus KG, Perlegas D, Hammarskjold ML, McNamara CA. Intron retention generates a novel Id3 isoform that inhibits vascular lesion formation. J Biol Chem 2004;279:32897-903.
- Deed RW, Jasiok M, Norton JD. Attenuated function of a variant form of the helix-loop-helix protein, Id-3, generated by an alternative splicing mechanism. FEBS Lett 1996;393:113-6.
- Maeda Y, Tsuji K, Nifuji A, Noda M. Inhibitory helix-loop-helix transcription factors Id1/Id3 promote bone formation in vivo. J Cell Biochem 2004;93:337-44.
- Ellmeier W, Weith A. Expression of the helix-loop-helix gene Id3 during murine embryonic development. Dev Dyn 1995;203:163-73.
- Jen Y, Manova K, Benezra R. Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Dev Dyn 1996;207:235-52.
- Jen Y, Manova K, Benezra R. Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn 1997;208:92-106.
- Persengiev SP, Kilpatrick DL. The DNA methyltransferase inhibitor 5-azacytidine specifically alters the expression of helix-loop-helix proteins Id1, Id2 and Id3 during neuronal differentiation. Neuroreport 1997;8:2091-5.
- Moldes M, Boizard M, Liepvre XL, Feve B, Dugail I, Pairault J. Functional antagonism between inhibitor of DNA binding (Id) and adipocyte determination and differentiation factor 1/sterol regulatory element-binding protein-1c (ADD1/SREBP-1c) trans-factors for the regulation of fatty acid synthase promoter in adipocytes. Biochem J 1999;344:873-80.
- Moldes M, Lasnier F, Feve B, Pairault J, Djian P. Id3 prevents differentiation of preadipose cells. Mol Cell Biol 1997;17:1796-804.
- Meyer KB, Skogberg M, Margenfeld C, Ireland J, Pettersson S. Repression of the immunoglobulin heavy chain 3' enhancer by helix-loop-helix protein Id3 via a functionally important E47/E12 binding site: implications for developmental control of enhancer function. Eur J Immunol 1995;25:1770-7.
- Alani RM, Young AZ, Shifflett CB. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc Natl Acad Sci USA 2001;98:7812-6.
- Nickoloff BJ, Chaturvedi V, Bacon P, Qin JZ, Denning MF, Diaz MO. Id-1 delays senescence but does not immortalize keratinocytes. J Biol Chem 2000;275:27501-4.
- Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003;115:281-92.
- Wilson JW, Deed RW, Inoue T, Balzi M, Becciolini A, Faraoni P, et al. Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res 2001;61:8803-10.
- Vandeputte DA, Troost D, Leenstra S, Ijlst-Keizers H, Ramkema M, Bosch DA, et al. Expression and distribution of Id helix-loop-helix proteins in human astrocytic tumors. Glia 2002;38:329-38.
- Langlands K, Down GA, Kealey T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res 2000;60:5929-33.
- Deleu S, Savonet V, Behrends J, Dumont JE, Maenhaut C. Study of gene expression in thyrotropin-stimulated thyroid cells by cDNA expression array: ID3 transcription modulating factor as an early response protein and tumor marker in thyroid carcinomas. Exp Cell Res 2002;279:62-70.
- Arnold JM, Mok SC, Purdie D, Chenevix-Trench G. Decreased expression of the Id3 gene at 1p36.1 in ovarian adenocarcinomas. Br J Cancer 2001;84:352-9.
- Sablitzky F, Moore A, Bromley M, Deed RW, Newton JS, Norton JD. Stage- and subcellular-specific expression of Id proteins in male germ and Sertoli cells implicates distinctive regulatory roles for Id proteins during meiosis, spermatogenesis, and Sertoli cell function. Cell Growth Differ 1998;9:1015-24.
- Albanese JM, Reuter VE, Bosl GJ, Houldsworth J, Chaganti RS. Expression of ID genes in differentiated elements of human male germ cell tumors. Diagn Mol Pathol 2001;10:248-54.
- Damdinsuren B, Nagano H, Kondo M, Yamamoto H, Hiraoka N, Yamamoto T, et al. Expression of Id proteins in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Int J Oncol 2005;26:319-27.
- Alway SE, Degens H, Krishnamurthy G, Chaudhrai A. Denervation stimulates apoptosis but not Id2 expression in hindlimb muscles of aged rats. J Gerontol A Biol Sci Med Sci 2003;58:687-97.
- Gundersen K, Merlie JP. Id-1 as a possible transcriptional mediator of muscle disuse atrophy. Proc Natl Acad Sci USA 1994;91:3647-51.
- Chen H. Gene expression by the anterior pituitary gland: effects of age and caloric restriction. Mol Cell Endocrinol 2004;222:21-31.
- Tzeng SF, Kahn M, Liva S. de VJ. Tumor necrosis factor-alpha regulation of the Id gene family in astrocytes and microglia during CNS inflammatory injury. Glia 1999;26:139-52.
- Shimizu-Nishikawa K, Tazawa I, Uchiyama K, Yoshizato K. Expression of helix-loop-helix type negative regulators of differentiation during limb regeneration in urodeles and anurans. Dev Growth Differ 1999;41:731-43.
- Wang X, Bhattacharyya D, Dennewitz MB, Kalinichenko VV, Zhou Y, Lepe R, et al. Rapid hepatocyte nuclear translocation of the Forkhead Box M1B (FoxM1B) transcription factor caused a transient increase in size of regenerating transgenic hepatocytes. Gene Expr 2003;11:149-62.
- Lopez-Carballo G, Moreno L, Masia S, Perez P, Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem 2002;277:25297-304.
- Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 2003;11:915-26.
- Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol 2004;24:4241-54.
- Peng Y, Kang Q, Luo Q, Jiang W, Si W, Liu BA, et al. Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J Biol Chem 2004;279:32941-9.
- Deed RW, Hara E, Atherton GT, Peters G, Norton JD. Regulation of Id3 cell cycle function by Cdk-2-dependent phosphoryla-tion. Mol Cell Biol 1997;17:6815-21.
- Hata K, Yoshimoto T, Mizuguchi J. CD40 ligand rescues inhibitor of differentiation 3-mediated G1 arrest induced by anti-IgM in WEHI-231 B lymphoma cells. J Immunol 2004;173:2453-61.
- Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat Immunol 2001;2:165-71.
- Mueller C, Baudler S, Welzel H, Bohm M, Nickenig G. Identification of a novel redox-sensitive gene, Id3, which mediates angiotensin II-induced cell growth. Circulation 2002;105:2423-8.
- Miyazono K, Miyazawa K. Id: a target of BMP signaling. Science STKE 2002; available from .http://stke.sciencemag.org/cgi/content/full/sigtrans;2002/151/pe40
- Yeh K, Lim RW. Genomic organization and promoter analysis of the murine Id3 gene. Gene 2000;254:163-71.
- Li XJ, Hata K, Mizuguchi J. Engagement of membrane immunoglobulin enhances Id3 promoter activity in WEHI-231 B lymphoma cells. Acta Pharmacol Sin 2005;26:486-91.
- Wu J, Lim RW. Regulation of inhibitor of differentiation gene 3 (Id3) expression by Sp2-motif binding factor in myogenic C2C12 cells: Down regulation of DNA binding activity following muscle differentiation. Biochim Biophy Acta 2005;1731:13–22.
- Nickenig G, Baudler S, Muller C, Werner C, Werner N, Welzel H, et al. Redox-sensitive vascular smooth muscle cell proliferation is mediated by GKLF and Id3 in vitro and in vivo. FASEB J 2002;16:1077-86.
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002;17:51-62.
- Wyzykowski JC, Winata TI, Mitin N, Taparowsky EJ, Konieczny SF. Identification of novel MyoD gene targets in proliferating myogenic stem cells. Mol Cell Biol 2002;22:6199-208.
- Berry FB, Miura Y, Mihara K, Kaspar P, Sakata N, Hashimoto-Tamaoki T, et al. Positive and negative regulation of myogenic differentiation of C2C12 cells by isoforms of the multiple homeodomain zinc finger transcription factor ATBF1. J Biol Chem 2001;276:25057-65.
- Brunner C, Laumen H, Nielsen PJ, Kraut N, Wirth T. Expression of the aldehyde dehydrogenase 2-like gene is controlled by BOB.1/OBF.1 in B lymphocytes. J Biol Chem 2003;278:45231-9.
- Deed RW, Jasiok M, Norton JD. Lymphoid-specific expression of the Id3 gene in hematopoietic cells. Selective antagonism of E2A basic helix-loop-helix protein associated with Id3-induced differentiation of erythroleukemia cells. J Biol Chem 1998;273:8278-86.
- Ehrlich M, Buchanan KL, Tsien F, Jiang G, Sun B, Uicker W, et al. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes. Hum Mol Genet 2001;10:2917-31.
- Eickhoff B, Ruller S, Laue T, Kohler G, Stahl C, Schlaak M, et al. Trichostatin A modulates expression of p21waf1/cip1, Bcl-xL, ID1, ID2, ID3, CRAB2, GATA-2, hsp86 and TFIID/TAFII31 mRNA in human lung adenocarcinoma cells. Biol Chem 2000;381:107-12.
- Eickhoff B, Germeroth L, Stahl C, Kohler G, Ruller S, Schlaak M, et al. Trichostatin A-mediated regulation of gene expression and protein kinase activities: reprogramming tumor cells for ribotoxic stress-induced apoptosis. Biol Chem 2000;381:1127-32.
- Plonczynski M, Hardy CL, Safaya S, Harrell A, McCoy L, Brinson A, et al. Induction of globin synthesis in K562 cells is associated with differential expression of transcription factor genes. Blood Cells Mol Dis 1999;25:156-65.
- Matsumura ME, Li F, Berthoux L, Wei B, Lobe DR, Jeon C, et al. Vascular injury induces posttranscriptional regulation of the Id3 gene: cloning of a novel Id3 isoform expressed during vascular lesion formation in rat and human atherosclerosis. Arterioscler Thromb Vasc Biol 2001;21:752-8.
- Chen B, Han BH, Sun XH, Lim RW. Inhibition of muscle-specific gene expression by Id3: requirement of the C-terminal region of the protein for stable expression and function. Nucleic Acids Res 1997;25:423-30.
- Barone MV, Pepperkok R, Peverali FA, Philipson L. Id proteins control growth induction in mammalian cells. Proc Natl Acad Sci USA 1994;91:4985-8.
- King PH, Levine TD, Fremeau RT Jr, Keene JD. Mammalian homologs of Drosophila ELAV localized to a neuronal subset can bind in vitro to the 3' UTR of mRNA encoding the Id transcriptional repressor. J Neurosci 1994;14:1943-52.
- Keene JD. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc Natl Acad Sci USA 2001;98:7018-24.
- Afouda AB, Reynaud-Deonauth S, Mohun T, Spohr G. Localized XId3 mRNA activation in Xenopus embryos by cytoplasmic polyadenylation. Mech Dev 1999;88:15-31.
- Bounpheng MA, Dimas JJ, Dodds SG, Christy BA. Degradation of Id proteins by the ubiquitin-proteasome pathway. FASEB J 1999;13:2257-64.
- Thatikunta P, Qin W, Christy BA, Tennekoon GI, Rutkowski JL. Reciprocal Id expression and myelin gene regulation in Schwann cells. Mol Cell Neurosci 1999;14:519-28.
- Liu CJ, Ding B, Wang H, Lengyel P. The MyoD-inducible p204 protein overcomes the inhibition of myoblast differentiation by Id proteins. Mol Cell Biol 2002;22:2893-5.
- Berse M, Bounpheng M, Huang X, Christy B, Pollmann C, Dubiel W. Ubiquitin-dependent degradation of Id1 and Id3 is mediated by the COP9 signalosome. J Mol Biol 2004;343:361-70.
- Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J 1998;17:5964-73.
- Fajerman I, Schwartz AL, Ciechanover A. Degradation of the Id2 developmental regulator: targeting via N-terminal ubiqui-tination. Biochem Biophys Res Commun 2004;314:505-12.
- Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol 2004;24:6140-50.
- Langlands K, Yin X, Anand G, Prochownik EV. Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J Biol Chem 1997;272:19785-93.
- Chen B, Lim RW. Physical and functional interactions between the transcriptional inhibitors Id3 and ITF-2b. Evidence toward a novel mechanism regulating muscle-specific gene expression. J Biol Chem 1997;272:2459-63.
- Deed RW, Armitage S, Norton JD. Nuclear localization and regulation of Id protein through an E protein-mediated chaperone mechanism. J Biol Chem 1996;271:23603-6.
- Stewart HJ, Zoidl G, Rossner M, Brennan A, Zoidl C, Nave KA, et al. Helix-loop-helix proteins in Schwann cells: a study of regulation and subcellular localization of Ids, REB, and E12/47 during embryonic and postnatal development. J Neurosci Res 1997;50:684-701.
- Nagata Y, Shoji W, Obinata M, Todokoro K. Phosphorylation of helix-loop-helix proteins ID1, ID2 and ID3. Biochem Biophys Res Commun 1995;207:916-26.
- Forrest ST, Taylor AM, Sarembock IJ, Perlegas D, McNamara CA. Phosphorylation regulates Id3 function in vascular smooth muscle cells. Circ Res 2004;95:557-9.
- Chaudhary J, Johnson J, Kim G, Skinner MK. Hormonal regulation and differential actions of the helix-loop-helix transcriptional inhibitors of differentiation (Id1, Id2, Id3, and Id4) in Sertoli cells. Endocrinology 2001;142:1727-36.
- Ishiguro A, Spirin KS, Shiohara M, Tobler A, Gombart AF, Israel MA, et al. Id2 expression increases with differentiation of human myeloid cells. Blood 1996;87:5225-31.
- Cooper CL, Brady G, Bilia F, Iscove NN, Quesenberry PJ. Expression of the Id family helix-loop-helix regulators during growth and development in the hematopoietic system. Blood 1997;89:3155-65.
- Tzeng SF. de VJ. Expression and functional role of the Id HLH family in cultured astrocytes. Brain Res Mol Brain Res 1997;46:136-42.
- Chaudhary J, Schmidt M, Sadler-Riggleman I. Negative acting HLH proteins Id1, Id2, Id3, and Id4 are expressed in prostate epithelial cells. Prostate 2005;64:353-64.
- Pammer J, Reinisch C, Kaun C, Tschachler E, Wojta J. Inhibitors of differentiation/DNA binding proteins Id1 and Id3 are regulated by statins in endothelial cells. Endothelium 2004;11:175-80.
- Shepherd TG, Nachtigal MW. Identification of a putative autocrine bone morphogenetic protein-signaling pathway in human ovarian surface epithelium and ovarian cancer cells. Endocrinology 2003;144:3306-14.
- Inuzuka H, Nanbu-Wakao R, Masuho Y, Muramatsu M, Tojo H, Wakao H. Differential regulation of immediate early gene expression in preadipocyte cells through multiple signaling pathways. Biochem Biophys Res Commun 1999;265:664-8.
- Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W, et al. Endostatin’s antiangiogenic signaling network. Mol Cell 2004;13:649-63.
- Watanabe T, Akishita M, Nakaoka T, He H, Miyahara Y, Yamashita N, et al. Caveolin-1, Id3a and two LIM protein genes are upregulated by estrogen in vascular smooth muscle cells. Life Sci 2004;75:1219-29.