
Protective effects of compound FLZ on β-amyloid peptide-(25–35)-induced mouse hippocampal injury and learning and memory impairment1
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that is clinically characterized by progressive memory loss and other cognitive impairments. The neuropathological hallmarks of AD include the formation of senile plaque of beta-amyloid (Aβ), neurofibrillary tangles, and synapse and neuron loss in several areas (hippocampus, cortex, amygdale, and others) of the brain[1]. Aβ has been thought to be a critical factor in the pathogenesis of AD. Many studies have found that the accumulation of Aβ in the brain is associated with progressive neuronal death and cognitive deficits[2–4]. Therefore, preventing the neurotoxicity induced by Aβ might be an optimal strategy for treatment of AD. Aβ25–35 is considered the shorter toxic fragment exerting neurotoxic effects similar to those produced by Aβ1–40/42, such as learning and memory impairment, neuronal apoptosis, cholinergic dysfunction, and oxidative stress[5,6], thus Aβ25–35 is usually used to establish the AD model for study of the neurotoxic properties of Aβ and drug screening.
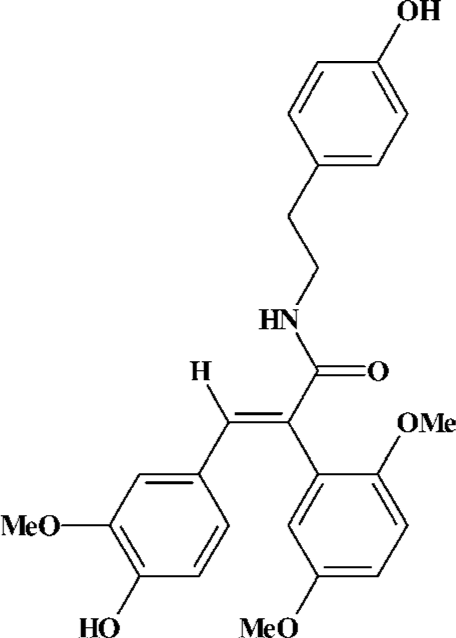
Because memory loss and cognitive dysfunction are the main clinical symptoms of AD patients, any therapy of AD requires identification of the factors that can confer protection against learning and memory impairment. Compound FLZ (Figure 1) is a novel synthetic cyclic analogue of natural squamosamide from a Chinese herb[7]. Our previous studies have demonstrated that compound FLZ has a strong antioxidant property, and that FLZ protected against the damage and apoptosis of primary cultured rat brain neurons and PC12 cells exposed to hydrogen peroxide, glutamate, N-methyl-D-aspartate (NMDA), dopamine, MPP+, and ischemia-reoxygenation. FLZ also improved abnormal behavior due to the functional disturbance of dopaminergic and cholinergic neurons in mice, indicating that FLZ possesses a neuroprotective property[8]. It is very interesting to study whether FLZ can attenuate learning and memory impairment, and pathological and biochemical damages in the hippocampus induced by icv injection of Aβ25–35 in mice.
Materials and methods
Reagents Compound FLZ was kindly provided by Professor Xiao-tian LIANG in the Department of Pharmaceutical Chemistry, Institute of Materia Medica, Chinese Academy of Medical Sciences. FLZ, a white powder with 99% purity, was suspended in 0.5% (w/v) sodium carboxymethyl cellulose (CMC-Na) for oral administration. Tacrine (Sigma Chemical, St Louis, MO, USA) was dissolved in saline. Aβ25–35 (Sigma) was dissolved in sterile double-distilled water at a concentration of 2 g/L, and incubated at 37 °C for 4 d for aggregation, and then stored at -20 °C for use[9]. Anti-acetylcholinesterase (AChE) polyclonal antibody was purchased from Boster Biotechnology (Boster, Wuhan, China). Bcl-2 and Bax polyclonal antibodies, second antibody and diaminobenzidine tetrahydrochloride (DAB) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other reagents were of analytical grade from Beijing Chemical Company (Beijing, China).
Animals Male ICR mice weighing 23–27 g were obtained from the Center of Experimental Animals, Chinese Academy of Medical Sciences (Grade II, Certificate N
Aβ25–35 injection and drug treatment The icv injection of Aβ25–35 was performed as described in a previous study[6]. Mice were lightly anesthetized with ether. The aged Aβ25–35 or saline at a volume of 7.5 µL was gradually injected into the right ventricle by a 28-gauge stainless-steel needle, with the following stereotaxic coordinates (in mm) from the bregma: A: -0.22, L: 1.0, V: 2.5. The injection point had already been confirmed at ventricle by injection of Indian ink in stead of Aβ25–35 peptide in preliminary experiments. The day after icv injection, mice were randomly divided into groups including: control, Aβ25–35 model, FLZ (75 mg/kg, 150 mg/kg), and tacrine (15 mg/kg). The FLZ, tacrine and vehicle (0.5% CMC-Na) were administered by gavage to mice once a day for 16 d. To observe the memory and learning function of mice, the step-down test was started on d 8 and the Morris water maze test on d 11 after the injection of Aβ25–35. The learning and memory tests were carried out between 9:00 AM and 18:00 PM.
Step-down test Amnesia in mice was examined through the step-down test on d 8 after the Aβ25–35 peptide injection. The apparatus (Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China) was a transparent acrylic cage . An insulated platform was fixed in the center of the grid floor. During the training, each mouse was kept in the cage for 3 min to adapt to the environment before electricity was delivered to the grid floor for 5 min. The retention test was carried out 24 h after the above training test. The grid floor was delivered with 36 v electricity, individual mice were placed on the insulated platform and the step-down latency and the number of errors within 5 min was recorded.
Morris water maze test The apparatus of Morris water maze (Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China) test is a white circular pool, 120 cm diameter and 40 cm deep, randomly divided into 4 quadrants. Milk powder was put into the pool to render the water opaque. The water temperature was maintained at 23±1 °C. A transparent Plexiglas platform was placed 2 cm below the water surface in the middle of one quadrant. The position of the platform was unchanged during the training trials. Two training trials per day were conducted for 5 consecutive days from d 11 after the injection of Aβ25–35. In each trial, a mouse was placed in the water faced to the pool wall at one starting position. The latency to find the platform was recorded up to 2 min. Mice that found the platform were allowed to remain on the platform for 30 s, and were then returned to the home cage. If the mouse could not find the platform within 2 min, it was gently guided to find the platform by the experimenter, and allowed to remain on the platform for 30 s, and the latency was recorded for 2 min. Data of each mouse behavior were collected by a video camera linked to a computer through an image analyzer. The total sum of latency in two trials of each mouse was counted as the individual result of a mouse per day. The mean latency was calculated by all tested mice per group per day.
At the end of the training period, mice were tested on a spatial probe trial in which the platform was removed, and they were allowed to swim freely for 2 min. The latency of the location of first crossing the platform and the number of crossings of the platform were recorded.
Malondialdehyde (MDA) measurement After completion of the Morris water maze behavior observation, 9–10 mice in every group were killed by decapitation. The hippocampus of the brain was rapidly removed and placed on ice. The dissected tissue was weighed and homogenized with cold 0.9% physiological saline. MDA content was determined by the thiobarbituric acid method. 1,1,3,3-tetra-methoxypropane was used as a standard[10].
Pathology examination of brain tissues After completion of the above behavior tests, 4 mice in every group were anesthetized with pentobarbital sodium (50 mg/kg, ip), and the brains were transcardially perfused with phosphate buffer solution (PBS, pH 7.4), followed by 4% paraformaldehyde in PBS (pH 7.4). The brains were removed and kept overnight in PBS containing 4% paraformaldehyde at 4 °C, and embedded in paraffin. Sections of 5-µm thickness were stained with hematoxylin and eosin. The lesions of brain tissues were observed with light microscope (NIKON E600, Japan) and the images were collected by image analysis system (Image Pro-Plus 7200, America, SONY 3CCD, Japan).
Immunohistochemistry assay of AChE, Bcl-2, and Bax After the brains were fixed as above, they were removed and kept overnight in PBS containing 20%–30% sucrose and 4% paraformaldehyde at 4 °C. Coronal sections of 35-μm thickness were cut using a cryostat at the hippocampus. Immunohistochemistry was performed as described in a previous study[11–13]. The primary antibodies were AChE (1:500), Bcl-2, and Bax (1:200). Immunostaining was visualized by the peroxidase method with a biotinylated anti-rabbit secondary antibody and diaminobenzidine oxidation (ABC kit, Santa Cruz). The primary antibody was replaced with normal serum in negative control.
Four mice were taken from each group for quantitative immunohistochemistry. One in every four sections was taken from a continuous series of sections prepared from hippocampal tissue. Six sections were selected in each mouse, so 24 sections of each group were read under a ×10 objective, and the number of positively stained cells in each group was counted. The mean of the number of positively stained cells was calculated from 24 sections of each group[11].
Statistical analysis Data are expressed as mean±SEM. Statistical analysis was performed by one-way ANOVA followed by least significant difference test. In all tests, P<0.05 was considered as statistically significant.
Results
Effect of FLZ on the learning and memory impairment induced by icv injection of Aβ25–35 in mice In the step-down test, the icv injection of aged Aβ25–35 (15 nmol/mouse) induced a significant decrease of the avoidance latency and increase of the number of errors, and also the step-down percentage of mice from the platform increased significantly (P<0.05). Oral administration of FLZ (75 mg/kg, 150 mg/kg) and tacrine (15 mg/kg) significantly improved the above impairments of learning and memory induced by Aβ25–35 in mice (P<0.05, Table 1). The latency of FLZ treated groups was restored to near that of the normal control group. The number of errors and step-down percentage were also improved markedly.

Full table
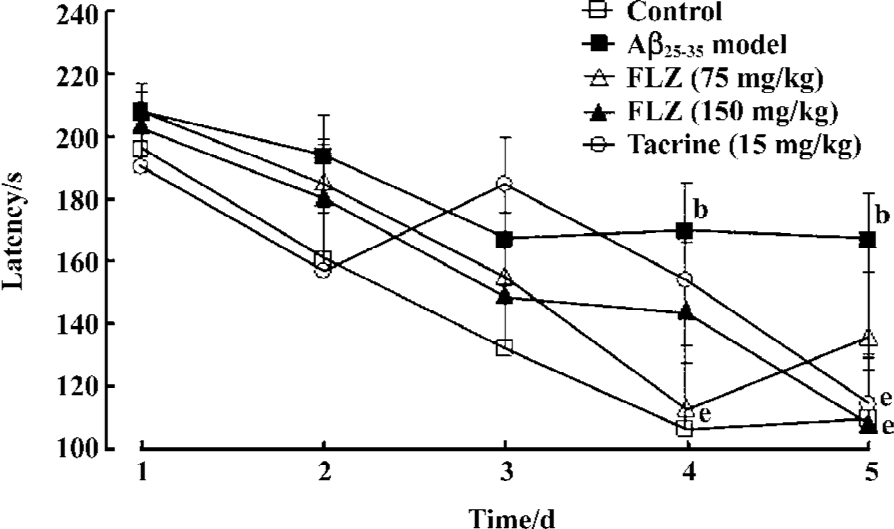
Furthermore, the Morris water maze was used to test the spatial learning and memory of each mouse from d 11 till d 16 after the icv injection of Aβ25–35. There was no difference of the mean latency to find the platform for mice between all groups on the first training day (d 11 after the icv injection of Aβ25–35). On the second day, the latency of mice in β-amyloid injected model group was longer than that of the control group, and still significantly longer on d 4 and d 5 (P<0.05). In comparison with Aβ25–35 model group, the latency of mice treated with FLZ (75 mg/kg, 150 mg/kg) significantly shortened on d 4 and d 5 (P<0.05). The latency of mice treated with tacrine also significantly decreased on d 5 (P<0.05, Figure 2).
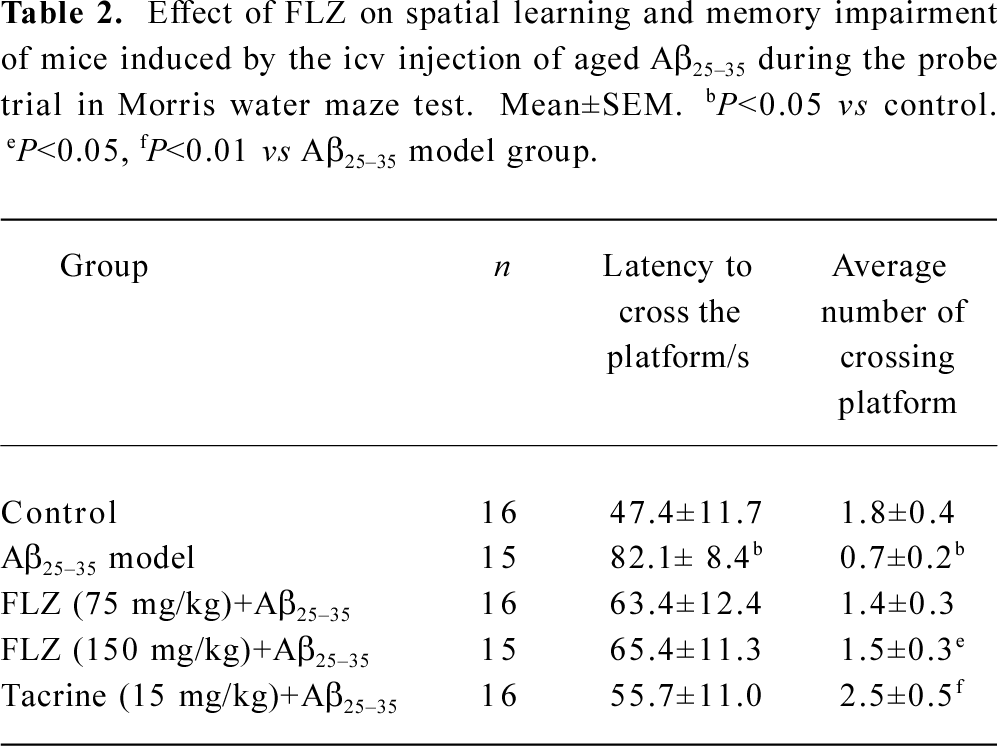
The spatial probe trial was performed to examine whether the mice had remembered the position of the platform. In comparison with the control group, the latency of location for first crossing the platform for mice in the Aβ25–35 model group increased (P<0.05), while the number of crossings of the platform for mice decreased (P<0.05). The treatment of mice with FLZ (75 mg/kg, 150 mg/kg) and tacrine (15 mg/kg) significantly shortened the latency of first-crossing the platform and increased the number of crossings of the platform within 2 min (P<0.05, Table 2).
Full table
All the results of the above two behavioral tests indicated that FLZ protected against the impairment of learning and memory function of mice induced by Aβ25–35.
Effect of compound FLZ on the MDA level in the hippocampus of mice icv injected with Aβ25–35 After completing the behavior test of Morris water maze test, the MDA level in the cerebral hippocampus of 9–10 mice per group was deter-mined. The icv injection of Aβ25–35 induced a significant increase in MDA level in the hippocampus (P<0.05). The treatment with FLZ (75 mg/kg, 150 mg/kg) and tacrine (15 mg/kg) markedly decreased MDA levels in the hippocampus of Aβ25–35-treated mice (Table 3).

Full table
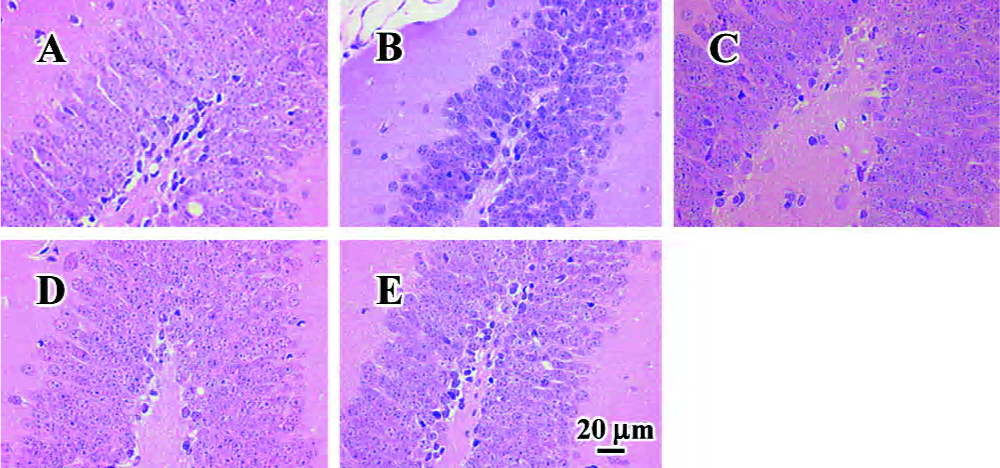
Effect of FLZ on the pathological injury of the hippocampus induced by icv injection of Aβ25–35 in mice No remarkable neuronal abnormalities in the hippocampus from mice of the normal control group were observed, while all examined brains of Aβ25–35 model group mice showed degeneration of neurons in the hippocampus and disorder of the array of neurons. The body of neuron became short and deeply stained with dye. Some neurons were shrunk and necrosed. Whereas the neurons in the FLZ (75 mg/kg, 150 mg/kg) and tacrine (15 mg/kg) group mice were close to that of normal control group, indicating that FLZ protected against the injuries of the hippocampus induced by Aβ25–35 injection (Figure 3).
Effect of FLZ on AChE, Bcl-2, and Bax immunoreactivity in the hippocampus cells of mice icv injected with Aβ25–35 There were few AChE and Bax immunoreactive neuronal cells in the hippocampal CA1 region of control group mice (Figure 4, 5). The staining was light and the immunoreactive cells had few processes. In the hippocampal CA1 region of Aβ25–35-icv injected mice, the number of AChE (P<0.05) and Bax (P<0.01) immunoreactive cells increased significantly in comparison with control group mice. The immunoreactive cells had long and darkly stained processes. Compared with Aβ25–35-treated mice, the number of AChE and Bax immunoreactive cells in the hippocampal CA1 region of FLZ (150 mg/kg) and tacrine-treated group mice was significantly reduced (P<0.05 or P<0.01), the staining was light, and the marked immunoreactive cells had few processes.
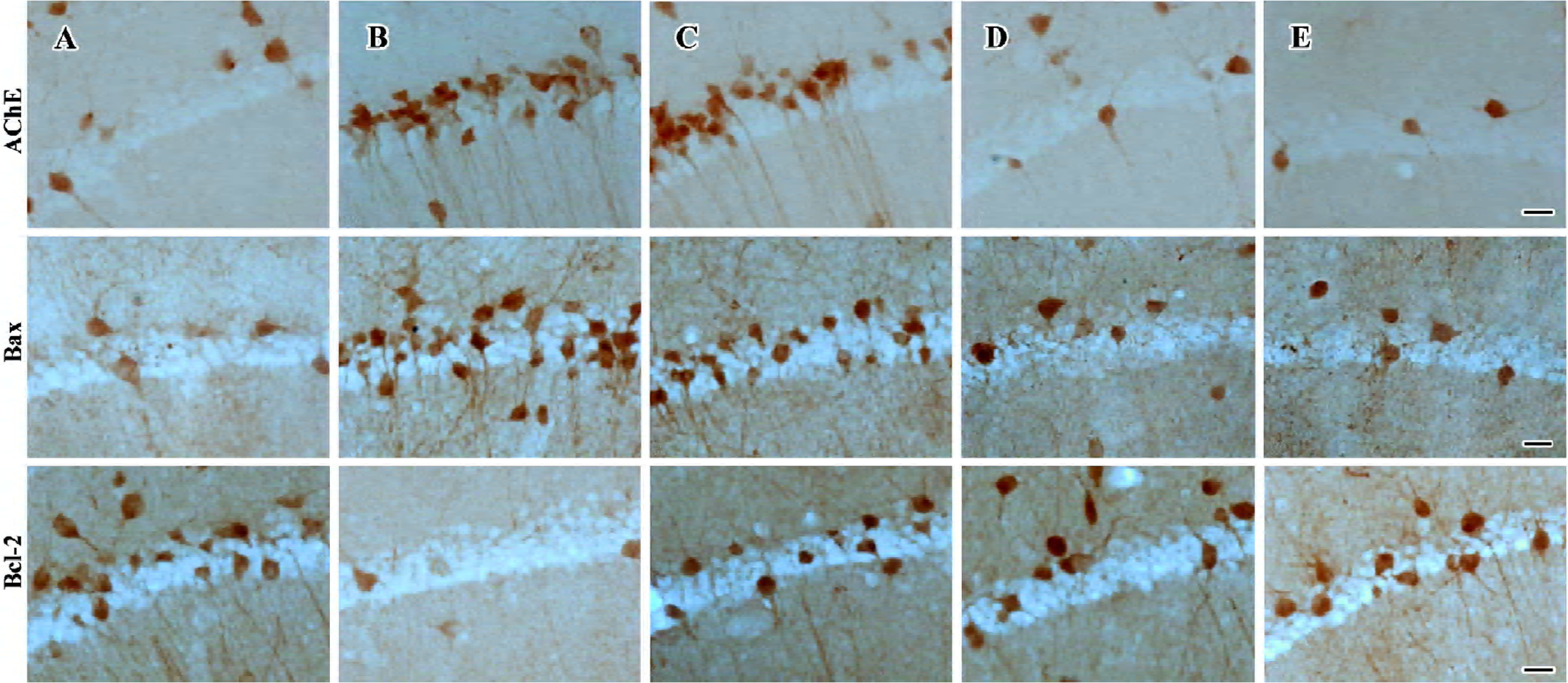
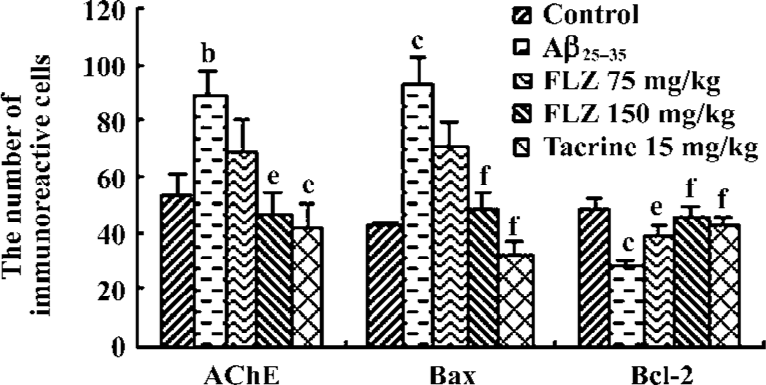
The results of Bcl-2 immunochemical staining showed that the hippocampal CA1 region of control group mice contained abundant and darkly stained Bcl-2 immunoreactive cells (Figure 4, 5). The marked cells had long processes. In the hippocampal CA1 region of Aβ25–35-treated mice, the number of Bcl-2 immunoreactive cells was less than that of control group mice (P<0.05), the staining was light, and the marked cells had few processes. There were more Bcl-2 immunoreactive cells in the hippocampal CA1 region of FLZ (150 mg/kg) and tacrine-treated mice in contrast to Aβ25–35-treated mice (P<0.01), and the immunoreactive cells had long processes. The staining was near to that of control group mice.
Discussion
Animal models are playing a critical role in ongoing attempts to understand the pathology and screen therapeutics of AD. Although no available model can meet all the full pathologic spectrum of AD disease, the injection of Aβ into the brain was shown to impair learning and memory, and elicit a degree of Alzheimer’s-type neurodegeneration[2–6]. The important point of Aβ25–35-induced amnesia is the influence of the physical state of the peptide, aggregated or soluble, at the time of administration. The in vitro incubation of Aβ25–35 leads to the formation of stable oligomeric aggregates, which contain an increased proportion of β-sheet structure that appears to be an important feature of the β-amyloid-induced neurotoxicity. This aggregation mimics in vitro the slow aging process that in vivo leads to the formation of the senile plaques[14,15]. In the present study, a single icv injection of aged Aβ25–35 to mice induced a significant impairment of learning and memory in step-down test and Morris water maze test, and obvious pathological lesions of the hippocampus. In mammals, the hippocampus is a critical neural structure in the early stage of memory formation. The impairment of memory formation is caused by damage in the hippocampus and associated areas of the temporal cortex. The deposition of Aβ first forms in temporal cortical regions including the hippocampus[16]. Some researchers reported that Aβ-induced injury of the hippocampus was associated with impairment in learning and memory beside biochemical changes and neuronal degeneration[12,17,18]. Compound FLZ was shown to prevent the neurotoxicity of Aβ25–35, as it significantly attenuated the learning and memory impairment and pathological injury of hippocampus in mice injected with Aβ25–35.
Apoptosis is considered as the main cause of the loss of cholinergic neurons in AD. Several authors reported that Aβ25–35 induced the neuronal degeneration through an apoptosis pathway in vitro and in vivo[19,20]. Bcl-2 family proteins play a pivotal role in regulating apoptotic cell death, some of which members, such as Bcl-2 and Bcl-xL, inhibit apoptosis and others such as Bax induces cell apoptosis. The relative ratio of proapoptotic and antiapoptotic proteins is important to determine cell survival or death[21]. The authors found that Aβ25–35 increased the expression of proapoptotic protein Bax and decreased the expression of anti-apoptotic protein Bcl-2 in the CA1 region of hippo-campus, indicating that Aβ25–35-induced pathological injury of the hippocampus was related to cell apoptosis. Compound FLZ exerted a significant anti-apoptotic effect through modulating the expression of Bcl-2 and Bax protein in the CA1 region of the hippocampus, which resulted in protection against pathological injury of the hippocampus and impairment of learning and memory.
Several studies with AD models demonstrated that Aβ increases AChE expression and AChE activity not only in cell culture but also in the intact brain[22,23]. AChE may promote beta-amyloid plaque formation and also increase the Aβ toxicity as reported by Rees et al[24]. Also, there is evidence to indicate that AChE is an exacerbating factor in the apoptosis of neurons, and plays a key role in the procession of apoptotic cell death. The apoptotic neurons can then secrete higher levels of AChE into the brain. This cascade amplification leads to progressive neuronal loss, which is the hallmark of AD[25,26]. In addition, cholinergic system dysfunction of AD is correlated with cognitive impairment. It was reported that acetylcholine (ACh) levels in the cerebral cortex and hippocampus were significantly decreased by centrally administered Aβ in animals[9,27]. AChE is a key enzyme in the catabolic metabolism of ACh. Aβ25–35-enhanced AChE expression and AChE activity might decrease the ACh level. In the present study, the icv injection of Aβ25–35 resulted in a significant increase of AChE expression, and compound FLZ markedly attenuated the increase of the number of AChE immunopositive cells in the CA1 region of the mouse hippocampus induced by icv injection of Aβ25–35. So, we considered that the inhibition of FLZ on AChE expression may be involved in anti-apoptosis and improvement of amnesia in mice injected with Aβ25–35. We also speculated that the inhibitory effect of FLZ on AChE expression might lead to an increase in acetylcholine level through inhibition of acetylcholine hydrolysis, which thereby enhanced the cholinergic function and improved the impairment of learning and memory. To confirm this speculation, direct measurement of hippocampal acetylcholine level and AChE activity is needed in future experiments.
Multiple lines of evidence indicated that oxygen free radicals (ROS) were involved in Aβ-induced neuronal apoptosis and death. Aβ can induce intracellular ROS production, which causes peroxidation of protein and lipid in neurons[28]. Aβ itself can transform into a radical state and further interacts with neuronal membranes[29]. MDA is an end product of lipid peroxidation of biomembranes, and the MDA content usually reflects the level of lipid peroxidation and indirectly reflects the extent of injury. The icv injection of Aβ25–35 induced a significant increase of MDA content in the mouse brain hippocampus, indicating that Aβ25–35 induced lipid peroxidation of neurons. FLZ was shown to have obvious antioxidant property, and also protected against oxidative damage and apoptosis of neurons induced by many toxins[8]. In the present study, we confirmed that FLZ significantly decreased the MDA content in the hippocampus of Aβ25–35-treated mice, suggesting that FLZ has a protective action against oxidative damage of neurons induced by Aβ25–35. Some antioxidants such as vitamin E were reported to eliminate Aβ-induced apoptosis and slow the progression of AD[30,31]. Xiao et al reported that the protective action of huperzine A and B and tacrine (AChE inhibitor) against Aβ toxicity to PC-12 cells was not cholinesterase-dependent but might be through an antioxidant pathway[32]. So, it appears that therapeutic efforts aiming at the removal of free radicals formation or preventing their damage to related neurons may be beneficial in pharmacol-therapy of AD.
The reason why tacrine, an AChE inhibitor, was selected as a positive control in the present study should be made clear. This was because tacrine was the first drug approved to treat AD by the FDA in the USA[33], and tacrine was known to attenuate the impairment of learning and memory induced by icv injection of Aβ25–35[9]. Tacrine also showed neuroprotective action against Aβ25–35-induced oxidative injury and H2O2-induced apoptosis[32,34]. The present results showed that both tacrine and FLZ have similar neuroprotective effect on icv injection of the Aβ25–35 model in mice. However, tacrine has been known to cause liver toxicity in AD patients by approximately 50% and has peripheral side effects[35]. Compound FLZ of the studied dosages had no such effects in mice. The acute oral LD50 of FLZ was over 5 g/kg body, indicating FLZ is a low toxicity compound. Moreover, not like tacrine, FLZ is not an inhibitor of AChE activity[8].
In summary, FLZ significantly attenuated learning and memory deficits as well as pathological and biochemical injuries of hippocampus in mice induced by icv injection of aged Aβ25–35 through the neuroprotective pathway.
Acknowledgements
We thank Prof Shu-li SHENG and Jian-jun ZHANG for their kind suggestions on this work.
References
- Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991;6:487-98.
- Olariu A, Yamada K, Mamiya T, Hefco V, Nabeshima T. Memory impairment induced by chronic intracerebroventricular infusion of beta-amyloid (1–40) involves down regulation of protein kinase C. Brain Res 2002;957:278-86.
- Hashimoto M, Hossain S, Shimada T, Sugioka K, Yamasaki H, Fujii Y, et al. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J Neurochem 2002;81:1084-91.
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 1999;2:271-6.
- Olariu A, Tran MH, Yamada K, Mizuno M, Hefco V, Nabeshima T. Memory deficits and increase emotionality induced β-amyloid (25–35) are correlated with the reduced acetylcholine release and altered phorbol dibutyrate binding in the hippocampus. J Neural Transm 2001;108:1065-79.
- Tohda C, Tamura T, Konatsu K. Repair of amyloid beta (25–35)-induced memory impairment and synaptic loss by a Kampo formula, Zokumei-to. Brain Res 2003;990:141-7.
- Xie P, Jiao XZ, Liang XT, Feng WH, Wei HL, Liu GT. Synthesis and antioxiactivity of squamosamide cyclic analogs. Acta Acad Med Sin 2004;26:372-8. Chinese..
- Feng W, Wei H, Liu G. Pharmacological study of the novel compound FLZ against experimental Parkinson’s models and its active mechanism. Mol Neurobiol 2005;31:295-300.
- Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res 1996;706:181-93.
- Yagi K. A simple flurometric assay for lipoperoxide in blood plasma. Biochem Med 1976;15:212-6.
- Zhao YM, Pei JJ, Ji ZJ, Zhao ZW, Qian YY, Sheng SL. Effect of amyloid precursor protein in 17mer peptide on microtubule structure and tau protein hyperphosphorylation in hippocampal neurons of experimental diabetic mice. Neuroreport 2003;14:61-6.
- Zhao ZW, Wang PW, Song LN, Zhuang XM, Ji ZJ, Sheng SL. Protection of liquid extract of Jiunaoyizhi capsule in impairment of hippocampal neurons of aging mice induced by D-galactose. Acta Anatom Sin 2001;32:343-5. Chinese..
- Stephan A, Laroche S, Davis S. Generation of aggregated β-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J Neurosci 2001;21:5703-14.
- Pick CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxi-city. Brain Res 1991;563:311-4.
- Delobette S, Privat A, Maurice T. In vitro aggregation facilitates β-amyloid peptide-(25–35)-induced amnesia in the rat. Eur J Pharmacol 1997;319:1-4.
- Ball MJ, Fisman M, Hachinski V, Blume W, Fox A, Kral VA, et al. A new definition of Alzheimer’s disease: a hippocampus dementia. Lancet 1985;1:14-6.
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid β-peptides. J Neurosci Res 2000;60:65-70.
- Sun MK, Alkon DL. Impairment of hippocampal CA1 hetero-synaptic transformation and spatial memory by β-amyloid 25–35. J Neurophysiol 2002;87:2441-9.
- Zeng H, Cheng Q, Zhao B. Genistein ameliorates beta-amyloid peptide (25-35)-induced hippocampal neuronal apoptosis. Free Radic Biol Med 2004;36:180-8.
- Wang R, Zhang HY, Tang XC. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by β-amyloid protein-(1–40) in rat. Eur J Pharmacol 2001;421:149-56.
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 2003;15:691-9.
- Hu W, Gray NW, Brimijoin S. Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J Neurochem 2003;86:470-8.
- Bronfman FC, Moechars D, Van Leuven F. Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein london mutant transgenic mice. Neurobiol Dis 2000;7:152-68.
- Rees T, Hammond P, Soreq H, Younkin S, Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol Aging 2003;2:777-87.
- Calderon FH, Von Bernhard R, De Ferrari G, Luza S, Aldunate R, Inestrosa NC. Toxic effects of acetylchonlinesterase on neuronal and glial-like cells in vitro. Mol Psychiatry 1998;3:247-55.
- Yang L, He HY, Zhang XJ. Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci Res 2002;42:261-8.
- Choi SH, Park CH, Koo JW, Seo JH, Kim HS, Jeong SJ, et al. Memory impairment and cholinergic dysfunction by centrally administered Aβ and carboxyl-terminal fragment of Alzheimer’s APP in mice. FASEB 2001;15:1816-8.
- Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. Free Radic Res 2002;36:1307-13.
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, et al. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer’s disease. Proc Natl Acad Sci USA 1994;91:3270-4.
- Cherubini A, Martin A, Andres-Lacueva C, Di Iorio A, Lamponi M, Mecocci P, et al. Vitamin E levels, cognitive impairment and dementia in older persons: the In CHIANTI study. Neurobiol Aging 2005;26:987-94.
- Huang HM, Ou HC, Hsieh SJ. Antioxidants prevent amyloid peptide-induced apoptosis and alteration of calcium homeostasis in cultured cortical neurons. Life Sci 2000;66:1879-92.
- Xiao XQ, Wang R, Tang XC. Huperzine A and Tacrine attenuate β-amyloid peptide-induced oxidative injury. J Neurosci Res 2000;61:564-9.
- Freeman SE, Dawson RM. Tacrine: a pharmacological review. Progress in Neurobiology 1991;36:257-77.
- Wang R, Zhou J, Tang XC. Tacrine attenuates hydrogen peroxide-induced apoptosis by regulating expression of apoptosis-related genes in rat PC12 cells. Brain Res Mol Brain Res 2002;107:1-8.
- Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271:992-8.