
Oncogenesis of RON receptor tyrosine kinase: a molecular target for malignant epithelial cancers1
Introduction
Receptor tyrosine kinases (RTK) are transmembrane proteins with unique structural properties[1,2]. Currently, twenty families of RTK have been identified[2]. They all share a similar structure consisting of a large extracellular domain, a short transmembrane segment, and an intracellular component with intrinsic tyrosine kinase activity[1,2]. The typical examples of RTK are EGFR and MET[3,4]. Upon binding of a specific ligand, the corresponding RTK undergoes dimerization, resulting in autophosphorylation of tyrosine residues in various domains that activate the intrinsic tyrosine kinase activities[5]. By recruiting, interacting with, and catalyzing various intracellular signaling proteins, the activated RTK gives rise to multiple signaling cascades that lead to gene activation and biological responses[5,6]. The activation of RTK can also be achieved through ligand-independent mechanisms[1]. Mutation, deletion, truncation, chromosomal rearrangement, and translocation have been widely documented in abnormal RTK activation under various pathological conditions[1]. In certain human tumors, aberrant RTK expression and activation, such as increased expression of the EGFR family members in breast and lung cancers, are hallmarks of tumor progression[7]. Thus, RTK are potential targets for therapeutic intervention. The inhibition of RTK activities by specific inhibitors, such as Herceptin which blocks EGFR2 (Her2), significantly improves clinical outcomes in patients with breast or lung cancer[8].
The recepteur d’origine nantais (RON) receptor tyrosine kinase belongs to the MET proto-oncogene family[9]. Since its discovery in 1993[9], significant progress has been made to elucidate its biochemical and biological properties[10,11]. RON has been found to be essential in embryonic development and in tumor invasive/malignant phenotypes[10,12]. This review summarizes current knowledge of RON in cancer progression and its potential as a therapeutic target. It is believed that by determining the mechanisms of RON in cancer pathogenesis we should not only be able to understand the pathogenic roles of RTK in epithelial cancer progression, but also have valuable information about novel strategies to treat malignant cancers.
Biochemical properties of RON and macro-phage stimulating protein (MSP)
RON gene and protein Human RON cDNA was cloned in 1993 from a keratinocyte cDNA library[9]. It encodes a protein with characteristics of a receptor tyrosine kinase[9]. The RON gene contains 20 exons and 19 introns and resides on chromosome 3p21[13,14]. The murine homologue of RON, known as stem cell derived kinase(STK), was isolated from bone marrow cells[15]. The RON cDNA encodes 1400 amino acids, which is synthesized first as a single-chain precursor (pro-RON)[9]. Maturation occurs in the cell membrane resulting in a 180-kDa heterodimeric protein composed of a 40 kDa α-chain and a 150 kDa transmembrane β-chain with intrinsic tyrosine kinase activity[16,17]. The extracellular sequences of RON contain several domains that include an N-terminal semaphoring (sema) domain, followed by the plexin, semaphor-, and integrin (PSI) domain, and four immunoglobulin-like IPT domains[9]. RON gene transcripts are present in the liver, lung, brain, kidney, bone, adrenal glands, testis, and digestive tract[18]. Western and Northern blot analyses indicate that RON is primarily expressed in cells of epithelial origin such as colon, breast, and skin[9,18]. Fibroblasts do not express RON. Promoter analysis of the RON gene transcription showed that SP-1 sites are important for RON gene transcription in epithelial cells[19]. The turnover of the RON protein is regulated through a mechanism in which c-Cbl ubiquitin ligase binds to phosphorylated RON, leading to its endocytosis and subsequent degradation[20]. Develop-mentally, RON is required for normal embryogenesis. Complete disruption of the RON gene (knockout) leads to the death of mouse embryos in the early stages[12].
Structurally, RON is a member of the MET proto-oncogene family[21], a distinct subfamily of RTK[1]. Only two members of the MET family, MET and RON, exist as is evident from human genomic sequence analysis[22]. MET is the receptor for hepatocyte growth factor/scatter factor[23] and has a well-described role in tumor progression[3]. Proteins highly homologous to human RON have been identified in other species including mouse[15,24], chicken[25,26], xenopus[27], and puffer fish[28]. In avian erythroblastosis retrovirus S13 that causes sarcoma, erythroblastosis, and anemia in young chicks[29], an oncoprotein called V-Sea was identified in the viral genome[30]. V-Sea is a hybrid protein containing the chicken Sea kinase domain fused with viral envelope sequences[31,32]. These data suggest that RON is evolutionally conserved in different species.
MSP gene and protein The only ligand identified for RON is MSP[16,17], a serum-derived protein that belongs to the plasminogen-related growth factor family[33,34]. MSP was discovered during the study of macrophage spreading, migration, and phagocytosis[33]. Sequence analysis confirmed that MSP is an 80 kDa protein with a putative signal peptide followed by a pre-activation segment, four kringle domains, and a serine-protease-like domain with the substitution of three amino acids in the active sites[34]. Between the 4th kringle domain region and the serine-protease like domain is an amino acid sequence (Arg483-Val-Val-gly-gly) serving as the cleavage site[34]. Thus, MSP is synthesized first as a single-chain precursor (pro-MSP). The proteolytic cleavage of pro-MSP yields a two-chain (α/β) mature MSP. Receptor binding and crystal structural analyses proved that MSP β-chain binds to RON in an ‘enzyme-substrate’ mode[35,36].
MSP is evolutionally conserved in different species. The cDNAs encoding human, mouse, chicken, and xenopus MSP have been isolated[26,27,34,37]. Among various tissues examined, MSP mRNA expression is mainly detected in hepatocytes[33,34,38], suggesting that liver cells are the major sources of MSP. Recent studies have shown that MSP mRNA is also present in cells from the kidney and lung[39,40]. However, the significance of these findings remains to be determined.
MSP is constantly produced and circulated in blood at optimal concentration as a biologically inactive pro-MSP[41,42]. The amount of pro-MSP in serum is approximately 2 to 5 nmol/L[41], a concentration optimal for biological activities. Gene knockout studies have revealed that inactivation of the MSP gene in mice is not lethal[43]. Mice developed normally and had no visible phenotypic changes[43]. These results suggest that MSP is not required for embryonic development and growth.
Oncogenic potentials in human epithelial cancers
Altered RON expression in primary human cancers Systematic analysis of RON expression in normal tissues and cancer samples has not been studied, although immunohistochemical staining has been conducted in several types of cancers[44–48]. A significant variation by using different antibodies exists in interpretation of the obtained results. In general, RON is detectable in certain types of normal cells such as epithelial cells and tissue macrophages. Elevated RON expression has been found in breast, colon, lung, bladder, and ovarian cancers[44–48]. The incidence of overexpression ranged from 32.8% to 59% dependent on individual types of cancers. In breast tissues, RON is relatively low in normal cells and benign lesions (adenoma and papillomas), but highly expressed in 47% (35 out of 75 cases) of tumor specimens with different histotypic variants[44]. Increased RON expression was strongly correlated to phosphorylation status and invasive activity[44], suggesting that RON abnormality plays a role in the progression of human breast carcinomas to invasive-metastatic phenotypes. Moreover, RON overexpression was an independent predictor of distant relapse in breast cancer[46].
Another example of abnormal RON expression is in colorectal cancers. RON is moderately expressed in normal colorectal mucosa, but increased significantly in the majority of primary human colorectal adenocarcinoma samples (29 out of 49 cases)[45]. Accumulated RON is also constitutively active with autophosphorylation[45,49] as a result of high levels of RON protein accumulation[45]. These results indicate that the activated RON transduces signals that regulate tumorigenic activities of colon cancer cells.
Recently, altered RON expression has been found in bladder and ovarian carcinomas[47,48]. In bladder cancer samples, aberrant RON expression was positively associated with histological grade, large size, non-papillary contour, and tumor stage[47]. In primary human ovarian carcinoma samples[48], the majority (29 out of 53 cases) had increased RON expression with a mixture of cytoplasmic and membrane staining pattern. Interestingly, the abnormal accumulation of RON is often accompanied by overexpression of MET in ovarian cancer cells, indicating that co-expression of RON and MET confer a selective advantage to ovarian cancers cells and might promote tumor progression[48].
We have recently evaluated the status of RON in various normal tissues and primary cancer samples using tissue microarray using a highly specific monoclonal antibody (clone ID2)[50]. Standardized analysis of the obtained results indicates that RON is wildly expressed in various types of normal cells at various degrees. Significant changes of RON expression were observed in cancer samples derived from stomach, colon, lung, liver, breast, thyroid, kidney, skin, cervical, bladder, brain, and lymph nodes (unpublished data). These results provide valuable information about the expression levels of RON in different types of normal and tumor tissues with significant pathological implications. They also indicate that altered RON expression is a distinct feature in certain types of cancers that might have prognostic or diagnostic value.
Generation of RON variants and their activities Altered RON expression in cancer cells is often accompanied by the generation of RON variants through mRNA splicing, alternative initiation, and protein truncation[45,49,51], which is responsible for RON protein diversity. Currently, six RON variants have been obtained and designated as RONΔ170, 165, 160, 155, 110, and 55 based on calculated molecular mass (Table 1). Following are the general features of these variants.
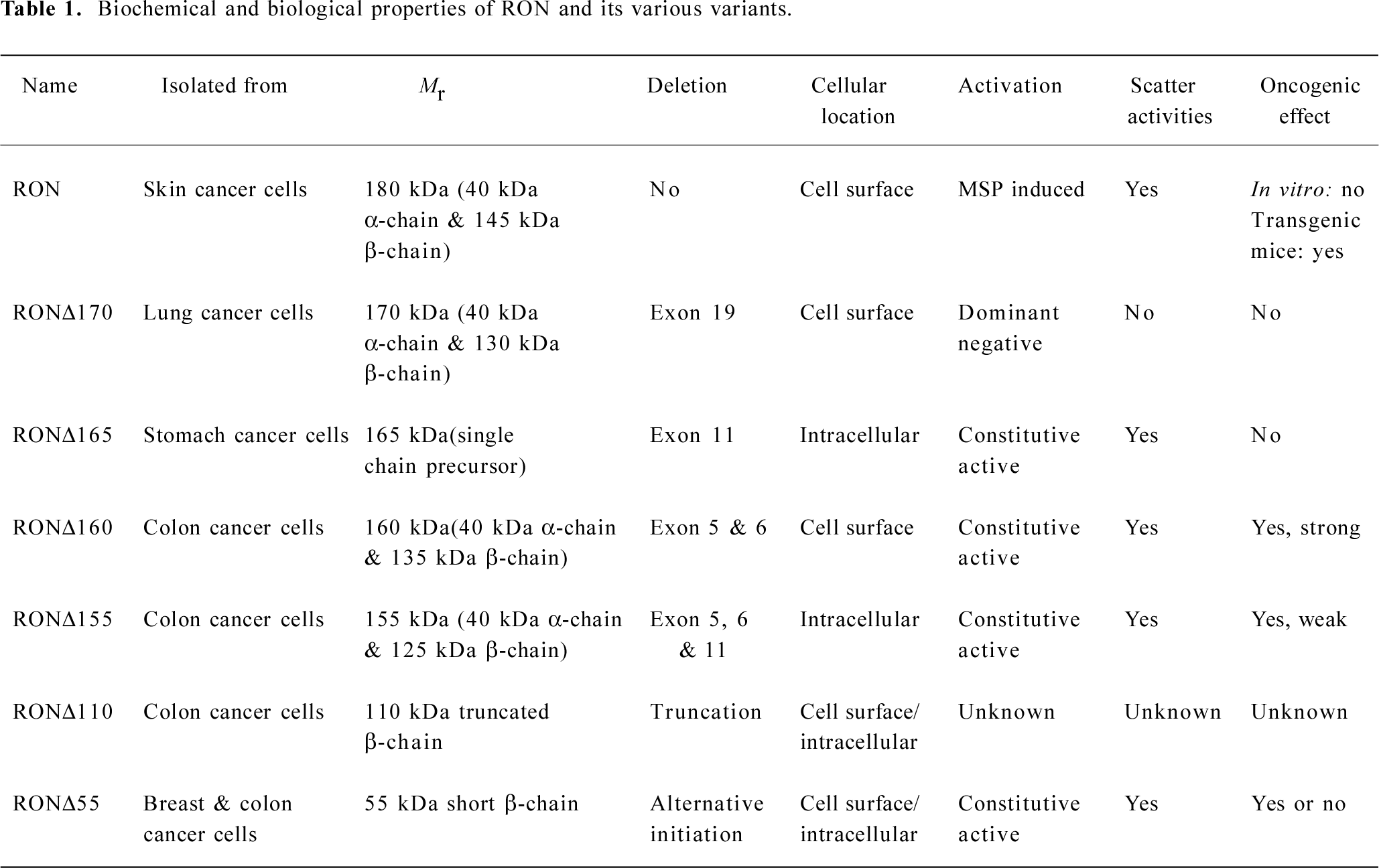
Full table
1) RONΔ170 is a variant with a deletion of 46 amino acids coded by exon 19 in the kinase domain[52]. The deletion also results in a reading-frame shift and creates a new stop code causing additional amino acid deletion. Preliminary results suggest that this protein is biologically inactive but acts as a dominant negative inhibitor (unpublished data).
2) RONΔ165 (also known as Δ-RON) was found in normal and malignant colon and breast tissues samples[45,53,54]. It has an in-frame deletion of 49 amino acids coded by exon 11 in the extracellular domain of the RON β-chain. The deletion prevents the proteolytic conversion of pro-RONΔ165 into the two-chain form and causes the protein to be retained in the cytoplasm. RONΔ165 does not have cell-transforming activities but is capable of inducing cell motile activities in transfected cells[45,53,54].
3) RONΔ160 was identified in primary CRC samples[45,51]. It has an in-frame deletion of 109 amino acids in the extracellular domain of the β-chain. These 109 amino acids are encoded by exons 5 and 6[13]. Unlike RONΔ165, the deletion of 109 amino acids does not affect the proteolytic processing of pro-RONΔ160. RONΔ160 is an oncogenic agent with constitutive kinase activities resulted from unbalanced numbers of cysteine residues in the extracellular domain of the RON β-chain[45,51].
4) RONΔ155 was cloned from two primary CRC samples[45]. It has a deletion of 158 amino acids coded by exons 5, 6, and 11 in the extracellular domain of the β-chain. RONΔ155 is constitutively active and capable of inducing tumor formation in vivo[45].
5) RONΔ110 is a truncated receptor discovered in certain colon cancer cell lines[49]. The protein is generated by proteolytic cleavage at Arg631-Lys632 residues. Thus, RONΔ110 is a variant lacking the RON-α chain and the most extracellular domain of the β-chain. We have recently constructed the RONΔ110 cDNA by molecular techniques and the function of RONΔ110 is currently under investigation (unpublished data).
6) The last variant is RONΔ55[55], also known as short form RON. The protein is produced by alternative initiation at Met913. It contains a short extracellular segment but retains the complete intracellular kinase domain and C-terminal tail. Studies from mice have indicated that RONΔ55 is critical in friend leukemia virus-induced erythroid proliferation and malignancy[56]. In human breast cancer cells, RONΔ55 modulate E-cadherin expression and thus might contribute to tumor progression[57]. Our studies have found that RONΔ55 does not have cell transforming activities but is capable of regulating cellular phenotypes (unpublished data). Thus, the roles of RONΔ55 in epithelial cancer pathogenesis remain to be determined.
It is currently unknown how RON variants are generated. DNA analysis indicates that all RON variants are not produced by abnormalities in the genomic sequences[45,49,51,53]. However, a recent study showed that skipping of exons such as exon 11 in RONΔ65 is controlled by a silencer and an enhancer of splicing located on exon 12[58]. Splicing factor SF2/ASF is responsible for the generation of RONΔ165 leading to increased invasive phenotypes[58]. Thus, splicing RON variants are generated through a complex mechanism that is critical to post-transcriptional regulation of RON expression and activation.
Tumorigenic activities of RON and its variants Wild-type (wt) RON has no cellular-transforming activities when expressed in NIH3T3 cells[59]. However, recent work showed that wtRON has the ability to mediate colony formation by immortalized human colonic AA/C1 epithelial cells[60]. The tumorigenic potentials of RON were also demonstrated by mutational studies[61]. Point mutations in the kinase domain such as D1232H/V or M1254T give RON the ability to cause cell transformation, tumor growth, and metastasis in nude mice[61–63]. Molecular modeling revealed that D1232H or M1254T substitution yieldes a dramatic increase in catalytic efficiency with oncogenic potential[62,63], suggesting that the single amino acid substitution favors the transition of the kinase from the inactive to the active state.
The oncogenic activities of two RON variants, RONΔ160 and RONΔ155 provide additional evidence indicating that RON is tumorigenic when its expression is altered[45]. Subcutaneous inoculation of NIH3T3 cells expressing RONΔ160 produced tumors in athymic nude mice and caused tumor metastasis to the lung[45]. Similarly, cells expressing RONΔ155 mediated tumor growth even though its efficiency is relatively low[45]. To our knowledge, these are the first naturally occurring RON variants identified to have tumor-producing activity.
A transgenic mouse model has also provided evidence indicating that RON overexpression can lead to tumor formation in vivo[64]. Overexpression of human RON in distal lung epithelial cells resulted in the formation of multiple lung adenomas and adenocarcinomas with unique cell morphology and growth pattern[64,65]. Tumors residing in peripheral portions of the lung appeared as solid-alveolar adenomas/adenocarcinomas and progressed slowly. Significant cellular atypia with a high mitotic index was observed in tumors at later stages. Some tumors showed distinct features of bronchioloalveolar carcinoma or large cell undifferentiated carcinoma with the pallor of the cytoplasm present in the tumors as well as the tremendous pleomorphism of cellular size and nuclear morphology[66]. Increased RON expression also led to genomic instability in tumor samples[64], suggesting that overexpression is the driving force leading to tumor initiation and progression in the lung distal epithelial cells in vivo.
RON-mediated invasive phenotypes RON-mediated cell motility including cell spreading, dissociation, migration, and matrix invasion are important functions essential for epithelial cell development and homeostasis[67]. These activities are also the hallmark of malignancy that distinguishes cancerous cells from benign tumors. Thus far, RON has been found to induce cell spreading, dissociation, migration, matrix invasion, and tubular formation in a variety of transformed or cancerous cells. Studies in vivo have further demonstrated that altered RON expression increases the metastatic potentials of tumors[61,62]. As shown in experimental lung metastasis experiments, NIH3T3 cells expressing RON mutants, such as M1254T or D1232V, display increased lung colonization activity[61]. Similarly, cells harboring splicing RON variants RONΔ160 or RONΔ150 have shown enhanced metastatic activities in athymic nude mice[45]. These results provide convincing evidence suggesting that RON-mediated invasive growth is manifested not only in cell culture but also in living animals.
Increased cellular invasiveness is often observed in a complicated event called epithelial-mesenchymal transition (EMT)[67,68], a distinct feature occurring during embryonic development and in tumor progression towards metastasis. RON has the ability to act independently or cooperate with other growth factors to induce EMT[69]. EMT is characterized by the loss of epithelial properties and the acquisition of mesenchymal phenotypes[67,68]. The typical EMT consists of the acquisition of a spindle-shaped morphology, delocalization of E-cadherin from cell junctions, elevated N-cadherin transcription, and expression of mesenchymal cellular markers such as β-smooth muscle actin[67,68]. Certain growth factors such as TGF-β or HGF/SF are known to induce EMT.
Using kidney epithelial cells as a model, we found that RON expression causes MDCK cell scattering with clear spindle-shaped morphology[69]. Cells also migrated through the collagen reconstituted basement membrane and penetrated into the Matrigel after MSP stimulation. Moreover, RON activation results in the redistribution of E-cadherin and re-expression of N-cadherin in spindle-shaped MDCK cells. These data suggest that RON activation directs a biochemical program that is indistinguishable from TGF-β1 or other growth factor-induced EMT. Additional studies further demonstrated that RON activation resultes in increased expression of the Smad 2 protein and directly causes its phosphorylation[69]. Smad 2 is a signal molecule responsible for TGF-β-induced biological activities including EMT[70]. The fact that RON activation mediates Smad 2 expression and phosphorylation suggests that RON-mediated EMT in epithelial cells might be channeled through the TGF-β/Smad signaling pathway. In support of this notion, we found that even though MSP and TGF-β are both capable of inducing EMT in MDCK cells, the complete epithelial-mesenchymal trans-differentiation (ie, expressing specific mesenchymal cellular markers such as α-smooth muscle actin) requires RON expression and activation[69]. In MDCK cells that do not express RON, neither TGF-β nor MSP alone is capable of inducing the expression of α-smooth muscle actin. However, when RON is expressed, TGF-β is able to induce α-smooth muscle actin expression[69]. These results suggest that RON expression is required for cell differentiation toward EMT with increased motile-invasive activities. As both MSP and TGF-β are involved in regulating epithelial tumor motility, the signaling collaborations between Smad 2 and RON might be essential in regulating invasive and metastatic potentials of certain epithelial cancers.
The signaling events of RON-mediated epithelial cell migration have been recently described using MSP-stimulated human keratinocytes as a model[71]. In quiescent keratinocytes, RON is physically associated with α3β1 integrin, but free from α6β4 integrin that is associated with hemidesmosome 1 and keratin filament leading to the formation of hemidesmosomes (structures supporting cell adhesion). MSP stimulates the RON receptor resulting in the activation of PI-3 kinase. The activated PI-3 kinase, through ATK kinase, phosphorylates both RON and α6β4 integrin at specific 14-3-3 binding sites. Subsequently, a protein complex is formed between RON and α6β4 integrin via 14-3-3 binding. This interaction prompts the relocation of α6β4 integrin from its original place at hemidesmosomes to lamellipodia. During these events, hemidesmosomes are gradually disassembled. The α3β1 integrin undergoes phosphorylation, is released from RON, and then moves to focal contacts. Concomitant with these changes, keratinocytes started to spread and migrate on laminin-5 coated membrane[71]. Thus, MSP induced epithelial cell migration is initiated through a mechanism in which RON forms a complex with α6β4 integrin in a 14-3-3 dependent manner, which results in activation of α3β1 integrin, disassembly of hemidesmosomes, and ultimately leads to cell spreading and migration.
Oncogenic signals Activated RON transduces a variety of signaling pathways[10]. Currently, signaling proteins are known to be activated by RON are SOS[72], Grb2[72], Ras[72], PI-3K[73,74], MAPK/Erk 1/2[61,63], JNK[49], β-catenin[75], FAK[76], integrins[77], Smad 2/3[69], and NF-κB complex[78]. We have recently found that β-arrestin-1, an adaptor protein for the seven transmembrane receptor family, is also involved in RON-mediated signaling events (unpublished data). These proteins are the effector molecules responsible for RON-mediated cell replication, transformation, migration, and matrix invasiveness.
Phosphorylation of tyrosine residues in the RON C-terminal tail results in the formation of a multifunctional docking site[76,79]. The docking site, also known as the bidentate motif, is composed of a conserved sequence encompassing two tyrosines (Y1353VQL-XXX-Y1360MNL-)[76,80]. Biochemical and biological studies have demonstrated that the docking site is critical in recruiting intracellular components such as PI-3K, Grb2, and others[79,80]. The substitution of Y1353 and Y1360 with other amino acids results in the significant impairment of the docking site in interacting with signaling proteins, which in turn leads to impaired cellular functions[80]. Mutational analysis has indicated that this docking site is involved in RON-mediated cell transformation. However, a recent study has shown that even though the docking site is required for the acquisition of the full oncogenic phenotype, certain RON mutants, such as RONM1254T, can exert cell transforming and metastatic activities without the docking site[80]. Consistent with these findings, a recent in vitro study has shown that the bidentate motif in the C-terminal tail inhibits RON kinase activities[81], suggesting that the C-terminal tail has a regulatory role that controls RON kinase catalytic activities.
As described above, the oncogenic potentials of RON are determined by the catalytic efficiency of the kinase activity. Currently, three mechanisms, overexpression, mutation, and truncation, are involved in the abnormal upregulation of the RON kinase activity. The overexpression model is documented in mouse lung epithelial cells, in which the accumulation of large amounts of wild-type RON results in constitutive activation of the kinase activity[64]. The formation of tumors in RON overexpressing lung epithelial cells, and not in control littermate mice[64], indicates that increased RON kinase activity could initiate unbalanced cell growth leading to tumor formation. The mutation model is observed in several experimentally created RON mutants, in which highly conserved residues in the RON kinase domain, such as D1232V or M1254T, are changed[61,63]. Substitution of these amino acid residues results in conformational changes in the kinase domain leading to a dramatic increase in kinase activity. Thus, oncogenic activities are easily acquired by such manipulation. The truncation model is observed in naturally occurring RON splicing variants such as RONΔ160 and RONΔ155[65]. The deletion of the particular extracellular regions encoded by exons 5, 6, and 11 results in increased kinase activity and tumorigenic activities. Detailed analysis of amino acid sequences have shown that this deletion causes an unbalanced number of cysteine residues in the extracellular domains of the RON variants resulting in the abnormal formation of intermolecular disulfide bonds. The consequence of such abnormal bond formation is oligomerization of the altered receptor leading to increased kinase activities, which are responsible for tumor formation in vivo.
Potential target for therapeutic intervention
Pathogenic activities of RON in tumor progression provide the basis for targeting RON for therapeutic intervention. In colon cancer cells, silencing the RON gene expression by siRNA techniques significantly affects cancer cell proliferation, soft agar growth, cellular motility, and increases apoptotic death[60]. Moreover, blocking RON expression greatly reduces Sw620 cell-mediated tumor growth in vivo[60]. These results clearly show that RON is critical for maintaining tumorigenic phenotypes. Thus, targeting RON expression should have a therapeutic potential to reverse malignant activities of colon cancers in vivo.
Various strategies have been developed to block RON expression and its activation. Experimentally, inhibition of MSP-induced activation by molecules that compete for MSP binding to RON is one approach. The soluble sema domain in the RON β chain extracellular sequences has been shown to inhibit MSP-induced RON activation and associated cancer cell proliferation[82]. The sema domain in the RON receptor is considered to be the MSP binding region. Another example is to use siRNA-mediated gene silencing to inhibit RON expression and its signaling events[60]. As described above, silencing RON gene expression significantly inhibits tumor formation and growth by colon cancer Sw620 cells in a nude mouse model. Therefore, RON-specific siRNA could have the potential to be delivered for therapeutic purposes. The third approach is to use a dominant negative agent to inactivate RON or its oncogenic variant. We have used RONΔ170 as a tool to block RON activation in cell models (unpublished data). RONΔ170 is a splicing variant defective in the kinase domain[52]. Preliminary studies indicate that this variant, when expressed in colon cancer cells, forms a heterodimeric complex with wtRON and oncogenic RONΔ160 leading to diminished receptor phosphorylation and signaling transduction (unpublished data). Thus, forced RONΔ170 expression as a gene therapy tool could have significance in reversing RON-mediated cancer-malignant phenotypes.
Realistic approaches with the most clinical usefulness include monoclonal antibodies (mAbs) and small molecule inhibitors. The development of a RON specific mAb is a very attractive idea. We have recently produced a panel of RON mAbs that specifically target the RON extracellular domain. Obtained results from in vitro studies have demonstrated that RON mAbs have the potential to regulate oncogenic activities mediated by RON or its variants (unpublished data). These studies pave the way for the development of RON mAb-based cancer therapy in the future.
Similar to other RTK such as EGFR, the RON tyrosine kinase is a candidate for small molecule inhibitors. Currently, small molecules that specifically inhibit RON kinase activities have not been reported. Because of structural similarities between MET and RON, selective small molecule inhibitors that target the kinase domain of both MET and RON have been obtained[83]. A recently described PHA665752 seems to meet this criterion. PHA665752 was originally discovered as a c-MET inhibitor with relatively improved potency and selectivity (cellular Met kinase: IC50, 0.042 µmol/L). It specifically blocks MET phosphorylation and associated tumor growth in mice in a dose-dependent manner[83]. Studies have also confirmed that PHA665752 targets RON kinase activities with a relative specificity (cellular RON kinase: IC50, 0.9 µmol/L)[83]. Thus, it is possible that RON-specific inhibitors could be identified through molecular designing.
Future direction
The progress in the last decade has dramatically increased our knowledge about the roles of RON in physiology and pathogenesis. The discovery of aberrant expression and activation in primary cancer samples indicates that RON is involved in the oncogenesis of epithelial tumors in vivo. Moreover, abnormal RON activation is critical in regulating malignant phenotypes of tumors. Thus, altered RON expression acts as a regulated invasiveness-promoting switch for the malignancy of certain epithelial cancers. From this point of view, it is important to determine the pathogenic roles of RON and its variants in the development of cancers such as colon and breast cancers. Furthermore, defining the relationships between the altered RON expression, including the variant forms of RON, and the observed colon cancer phenotypes is also worth pursuing. In this sense, mAb and small molecule inhibitor-based strategies to inactivate RON or its variants should provide important information about the pathological role of RON in epithelial cancers. Considering these facts, altered RON expression could be used as a cellular marker for early diagnosis and a pharmaceutical target for treatment of certain malignancies. Thus, understanding the roles of RON in tumorigenesis in lung or colon cancers offers an opportunity to uncover the molecular mechanisms underlying epithelial cancer pathogenesis. It should also hold potential for developing novel approaches to control oncogenesis of certain epithelial tumors in vivo.
Acknowledgements
The authors thank the colleagues in TTUHSC for their support and encouragement. We also thank Ms C SPIDEL for editing the manuscript.
References
- Robertson SC, Tynan JA, Donoghue DJ. RTK mutations and human syndromes: when good receptors turn bad. Trends Genet 2000;16:265-71.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912-34.
- Vande Woude GF, Jeffers M, Cortner J, Alvord G, Tsarfaty I, Resau J. Met-HGF/SF: tumorigenesis, invasion and metastasis. Ciba Found Symp 1997;212:119-30.
- Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology 2001;61:1-13.
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000;103:211-25.
- van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 1994;10:251-337.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70.
- Hortobagyi GN. Overview of treatment results with trastuzumab (Herceptin) in metastatic breast cancer. Semin Oncol 2001;6:43-7.
- Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene 1993;8:1195-202.
- Wang MH, Wang D, Chen YQ. Oncogenic and metastatic potentials of human macrophage stimulating protein receptor, the RON receptor tyrosine kinase. Carcinogenesis 2003;23:1291-300.
- Wang MH, Zhou YQ, Chen YQ. Macrophage stimulating protein and RON receptor tyrosine kinase: potential regulators of macrophage inflammatory activities. Scan J Immunol 2002;56:545-53.
- Muraoka RS, Sun WY, Colbert MC. The Ron/STK receptor tyrosine kinase is essential for peri-implantation development in the mouse. J Clin Invest 1999;103:1277-85.
- Angeloni D, Danilkovitch-Miagkova A, Ivanov SV, Breathnach R, Johnson BE, Leonard EJ, et al. Gene structure of the human receptor tyrosine kinase RON and mutation analysis in lung cancer samples. Genes Chromosomes Cancer 2000;29:147-56.
- Zabarovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 2002;21:6915-35.
- Iwama A, Okano K, Sudo T, Matsuda Y, Suda T. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood 1994;83:3160-9.
- Gaudino G, Follenzi A, Naldini L. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J 1994;13:3524-32.
- Wang MH, Ronsin C, Gesnel MC, Coupeym L, Skeel A, Leonard EJ, et al. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science 1994;266:117-9.
- Gaudino G, Avantaggiato V, Follenzi A, Acampora D, Simeone A, Comoglio PM. The proto-oncogene RON is involved in development of epithelial, bone and neuro-endocrine tissues. Oncogene 1995;11:2627-37.
- Del Gatto F, Gilbert E, Ronsin C, Breathnach R. Structure of the promoter for the human macrophage stimulating protein receptor gene. Biochim Biophy Acta 1995;1263:93-5.
- Penengo L, Rubin C, Yarden Y, Gaudino G. c-CBl is a critical modulator of the RON tyrosine kinase receptor. Oncogene 2003;22:3669-79.
- Rubin JS, Bottaro DP, Aaronson SA. Hepatocyte growth factor/scatter factor and its receptor, the c-met proto-oncogene product. Biochem Biophys Acta 1993;1155:357-71.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912-34.
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251:802-4.
- Wang MH, Iwama A, Skeel A, Suda T, Leonard EJ. The murine stk gene product, a transmembrane protein tyrosine kinase, is a receptor for macrophage-stimulating protein. Proc Natl Acad Sci USA 1995;92:3933-7.
- Huff JL, Jelinek MA, Borgman CA, Lancing TL, Parsons JT. The protooncogene c-sea encodes a transmembrane protein-tyrosine kinase related to the Met/hepatocyte growth factor/scatter factor receptor. Proc Natl Acad Sci USA 1993;90:6140-4.
- Wahl RC, Hsu RY, Huff JL. Chicken macrophage stimulating protein is a ligand of the receptor protein-tyrosine kinase Sea. J Biol Chem 1999;274:26361-8.
- Nakamura T, Aoki S, Takahashi T, Matsumoto K, Kiyohara T, Nakamura T. Cloning and expression of Xenopus HGF-like protein (HLP) and Ron/HLP receptor implicate their involvement in early neural development. Biochem Biophys Res Commun 1996;224:564-73.
- Cottage A, Clark M, Hawker K, Umrania Y, Wheller D, Bishop M, et al. Three receptor genes for plasminogen related growth factors in the genome of the puffer fish Fugu rubripes. FEBS Lett 1999;443:370-4.
- Bos TJ, Beug H, Graf T, Hayman M, Maki Y, Uckert W, et al. Two new retroviral onc genes, sea and jun. Princess Takamatsu Symp 1986;17:23-30.
- Smith DR, Vogt PK, Hayman MJ. The v-sea oncogene of avian erythroblastosis retrovirus S13: another member of the protein-tyrosine kinase gene family. Proc Natl Acad Sci USA 1989;86:5291-5.
- Agazie Y, Ischenko I, Hayman MJ. Concomitant activation of the PI3K-Akt and the Ras-ERK signaling pathways is essential for transformation by the V-SEA tyrosine kinase oncogene. Oncogene 2002;21:697-707.
- Hayman MJ, Kitchener G, Vogt PK, Beug H. The putative transforming protein of S13 avian erythroblastosis virus is a transmembrane glycoprotein with an associated protein kinase activity. Proc Natl Acad Sci USA 1985;82:8237-41.
- Skeel A, Yoshimura T, Showalter SD, Tanaka S, Appella E, Leonard EJ. Macrophage-stimulating protein: purification, partial amino acid sequence, and cellular activity. J Exp Med 1991;173:1227-34.
- Yoshimura T, Yuhki N, Wang MH, Skeel A, Leonard EJ. Cloning, sequencing, and expression of human macrophage stimulating protein (MSP, MST1) confirms MSP as a member of the family of kringle proteins and locates the MSP gene on chromosome 3. J Biol Chem 1993;268:15461-8.
- Wang MH, Montero-Julian FA, Breathnach R, Godowski PJ, Takehara T, Yoshikawa W, et al. Macrophage stimulating protein (MSP) binds to its receptor via the MSP beta chain. J Biol Chem 1997;272:16999-7004.
- Carafoli F, Chirgadze DY, Blundell TL, Gherardi E. Crystal structure of the beta-chain of human hepatocyte growth factor-like/macrophage stimulating protein. FEBS J 2005;272:5799-807.
- Degen FSJ, Stuart LA, Han S, Jamison CS. Characterization of the mouse cDNA and gene coding for a hepatocyte growth factor-like protein: expression during development. Biochemistry 1991;30:9781-91.
- Bezerra JA, Witte DP, Aronow BJ, Degen SJ. Hepatocyte-specific expression of the mouse hepatocyte growth factor-like protein. Hepatology 1993;8:394-9.
- Rampino T, Collesi C, Gregorini M, Maggio M, Soccio G, Guallini P, et al. Macrophage-stimulating protein is produced by tubular cells and activates mesangial cells. J Am Soc Nephrol 2002;13:649-57.
- Willett CG, Smith DI, Shridhar V, Wang MH, Emanuel RL, Patidar K, et al. Differential screening of a human chromosome 3 library identifies hepatocyte growth factor-like/macrophage-stimulating protein and its receptor in injured lung. Possible implications for neuroendocrine cell survival. J Clin Invest 1997;99:2979-91.
- Wang MH, Skeel A, Yoshimuram T, Copeland TD, Sakaguchi K, Leonard EJ. Antibodies to macrophage stimulating protein (MSP): specificity, epitope interactions, and immuno-assay of MSP in human serum. J Leukoc Biol 1993;54:289-95.
- Wang MH, Yoshimura T, Skeel A, Leonard EJ. Proteolytic conversion of single chain precursor macrophage stimulating protein to a biologically active heterodimer by contact enzymes of the coagulation cascade. J Biol Chem 1994;269:3436-40.
- Bezerra JA, Carrick TL, Degen JL, Witte D, Degen FSJ. Biological effects of targeted inactivation of hepatocyte growth factor-like protein in mice. J Clin Invest 1998;101:1175-83.
- Maggiora P, Marchio S, Stella MC, Giai M, Belfiore A, De Bortoli M, et al. Overexpression of the RON gene in human breast carcinoma. Oncogene 1998;16:2927-33.
- Zhou YQ, He C, Chen YQ, Wang D, Wang MH. Altered expression of the RON receptor tyrosine kniase in primary human colorectal adenocarcinomas: generation of different splicing variants and their oncogenic potential. Oncogene 2003;22:186-97.
- Lee WY, Chen HH, Chow NH, Su WC, Lin PW, Guo HR. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin Cancer Res 2005;11:2222-8.
- Cheng HL, Liu HS, Lin YJ, Chen HH, Hsu PY, Chang TY, et al. Co-expression of RON and MET is a prognostic indicator for patients with transitional-cell carcinoma of the bladder. Br J Cancer 2005;92:1906-14.
- Maggiora P, Lorenzato A, Fracchioli S, Costa B, Castagnaro M, Arisio R, et al. The RON and MET oncogenes are co-expressed in human ovarian carcinomas and cooperate in activating invasiveness. Exp Cell Res 2003;288:382-9.
- Chen YQ, Zhou YQ, Angeloni-Andreazzoli D, Kurtz AL, Qiang XZ, Wang MH. Overexpression and activation of the RON receptor tyrosine kinase in a panel of human colorectal carcinoma cells lines. Exp Cell Res 2000;261:229-38.
- Montero-Julian FA, Dauny I, Flavetta S, Ronsin C, Andre F, Xerri L, et al. Characterization of two monoclonal antibodies against the RON tyrosine kinase receptor. Hybridoma 1998;17:541-51.
- Wang MH, Kurtz AL, Chen YQ. Identification of a novel splicing product of the RON receptor tyrosine kinase in human colorectal carcinoma cells. Carcinogenesis 2000;21:1507-12.
- Angeloni D, Duh FM, Moody M, Dean M, Zabarovsky ER, Sentchenkom V, et al. C to A single nucleotide polymorphism in intron 18 of the human MST1R (RON) gene that maps at 3p21.3. Mol Cell Probes 2003;17:55-7.
- Collesi C, Santoro MM, Gaudino G, Comoglio PM. A splicing variant of the RON gene transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol 1996;16:5518-26.
- Okino T, Egami H, Ohmachi H, Takai E, Tamori Y, Nakagawa K, et al. Presence of RON receptor tyrosine kinase and its splicing variant in malignant and non-malignant human colonic mucosa. Intern J Oncol 1999;13:709-14.
- Nishigaki K, Thompson D, Hanson C, Yugawa T, Ruscetti S. The envelope glycoprotein of friend spleen focus-forming virus covalently interacts with and constitutively activates a truncated form of the receptor tyrosine kinase Stk. J Virol 2001;75:7893-903.
- Persons DA, Paulson RF, Loyd MR, Herley MT, Bodner SM, Bernstein A, et al. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat Genet 1999;23:159-65.
- Bardella C, Costa B, Maggiora P, Patane’ S, Olivero M, Ranzani GN, et al. Truncated RON tyrosine kinase drives tumor cell progression and abrogates cell-cell adhesion through E-cadherin transcriptional repression. Cancer Res 2004;64:5154-61.
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005;20:881-90.
- Santoro MM, Collesi C, Grisendi S, Gaudino G, Comoglio PM. Constitutive activation of the RON gene promotes invasive growth but not transformation. Mol Cell Biol 1996;16:7072-83.
- Xu XM, Wang D, Shen Q, Chen YQ, Wang MH. RNA-mediated gene silencing of the RON receptor tyrosine kinase alters oncogenic phenotypes of human colorectal carcinoma cells. Oncogene 2004;23:8464-74.
- Santoro MM, Penengo L, Minetto M, Orecchia S, Cilli M, Gaudino G. Point mutations in the tyrosine kinase domain release the oncogenic and metastatic potential of the Ron receptor. Oncogene 1998;17:741-9.
- Peace BE, Hughes MJ, Degen SJ, Waltz SE. Point mutations and overexpression of Ron induce transformation, tumor formation, and metastasis. Oncogene 2001;20:6142-51.
- Williams TA, Longati P, Pugliese L, Gual P, Bardelli A, Michieli P. MET(PRC) mutations in the RON receptor result in upregulation of tyrosine kinase activity and acquisition of oncogenic potential. J Cell Physiol 1999;181:507-14.
- Chen YQ, Zhou YQ, Fu LH, Wang D, Wang MH. Multiple pulmonary adenomas in the lung of transgenic mice overexpress-ing the RON receptor tyrosine kinase. Carcinogenesis 2002;23:1811-9.
- Zhou YQ, Chen YQ, Fisher JH, Wang MH. Targeted expression of the receptor tyrosine kinase RON in distal lung epithelial cells results in multiple tumor formation: oncogenic potential of RON in vivo. Oncogene 2002;21:6382-6.
- Barkley JE, Green MR. Bronchioloalveolar carcinoma. J Clin Oncol 1996;14:2377-86.
- Thiery JP, Chopin D. Epithelial cell plasticity in development and tumor progression. Cancer Metastasis Rev 1999;18:31-42.
- Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 2001;23:912-23.
- Wang D, Shen Q, Chen YQ, Wang MH. Collaborative activities of macrophage-stimulating protein and transforming growth factor-beta1 in induction of epithelial to mesenchymal transition: roles of the RON receptor tyrosine kinase. Oncogene 2004;23:1668-80.
- Miyazono K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev 2000;11:15-22.
- Santoro MM, Gaudino G, Marchisio PC. The MSP receptor regulates α6β4 and α3β1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell 2003;5:257-71.
- Li BQ, Wang MH, Kung HF. Macrophage-stimulating protein activates Ras by both activation and translocation of SOS nucleotide exchange factor. Biochem Biophys Res Commun 1996;216:110-8.
- Danilkovitch A, Skeel A, Leonard EJ. Macrophage stimulating protein-induced epithelial cell adhesion is mediated by a PI3-K-dependent, but FAK-independent mechanism. Exp Cell Res 1999;248:575-82.
- Wang MH, Montero-Julian FA, Leonard EJ. Requirement of PI-3 kinase for epithelial cell migration activated by human macrophage stimulating protein. Oncogene 1996;13:2167-75.
- Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol 2001;21:5857-68.
- Ponzetto C, Bardelli A, Zhen Z, Maina F. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994;77:261-71.
- Danilkovitch-Miagkova A, Angeloni D, Skeel A, Donley S, Lerman M, Leonard EJ. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. J Biol Chem 2000;275:14783-6.
- Zhou YQ, Chen YQ, Fisher JH, Wang MH. Activation of the RON receptor tyrosine kinase by macrophage-stimulating protein inhibits inducible cyclooxygenase-2 expression in murine macrophages. J Biol Chem 2002;277:38104-10.
- Xiao ZQ, Chen YQ, Wang MH. Requirement of both tyrosine kinase residues 1330 and 1337 in the c-terminal tail of the RON receptor tyrosine kinase for epithelial cell scattering and migration. Biochem Biophy Res Commun 2000;267:669-75.
- Santoro MM, Penengo L, Orecchia S, Cilli M, Gaudino G. The Ron oncogenic activity induced by the MEN2B-like substitution overcomes the requirement for the multifunctional docking site. Oncogene 2000;19:5208-11.
- Yokoyama N, Ischenko I, Hayman MJ, Miller WT. The C terminus of RON tyrosine kinase plays an autoinhibitory role. J Biol Chem 2005;280:8893-900.
- Angeloni D, Danilkovitch-Miagkova A, Miagkov A, Leonard EJ, Lerman MI. The soluble sema domain of the RON receptor inhibits macrophage-stimulating protein-induced receptor activation. J Biol Chem 2004;279:3726-32.
- Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res 2003;63:7345-55.