
CPU 86017, p-chlorobenzyltetrahydroberberine chloride, attenuates monocrotaline-induced pulmonary hypertension by suppressing endothelin pathway1
Introduction
Pulmonary arterial hypertension (PAH) is a common disease induced by many etiological factors. Pulmonary vascular remodeling by monocrotaline (MCT) affects both vascular smooth muscle and vascular wall connective tissue at the molecular level[1]. Endothelium plays an important role in controlling vascular tone as well as vascular remodeling by releasing endothelium-derived relaxing factors [EDRF, nitric oxide (NO)] and contracting factors mainly endothelin-1 (ET-1)[2]. ET-1 is a potent endothelium-derived vasoconstrictor peptide, which possesses mitogenic effects to proliferate the vascular smooth muscle cells (VSMC) in the development of PAH[3]. NO, which is released from the endothelium, attenuates proliferation of VSMC[4] and is opposed functionally to ET-1[5].
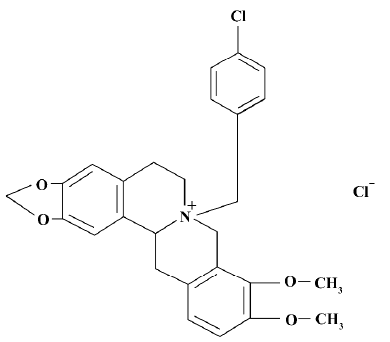
Monocrotaline (MCT), a pyrrole alkaloid, causes substantial and sustained inflammatory damage to the pulmonary arterial system and, therefore, causes PAH in rats[5]. CPU 86017, p-chlorobenzyltetrahydroberberine (Figure 1), a new compound derived from berberine, possesses a calcium channel blocking effect[6], combined with an antioxidative effect[7]. CPU 86017 ameliorates the progression of chronic congestive heart failure in relation to suppression of the ET-1 system[8]. Therefore, it is hypothesized that CPU 86017 might relieve the pulmonary hypertension, with an indirect effect on the overactive ET pathway underlying the pulmonary hypertension induced by MCT. We intend to explore the pathogenesis of PAH induced by MCT in relation to an activated ET pathway, and its downstream events (iNOS and ROS activity) in response to the medication of CPU 86017.
Materials and methods
Experimental protocol Sixty-five male Sprague-Dawley rats (weighing 250 g-300 g, from the Experimental Animal Center of Nanjing Medical University) were randomized into 5 groups (n =13). Animals (apart from the normal group) were injected with a single dose 60 mg/kg, sc) of MCT (Promega). p-Chloro-benzyl-tetrahydro-berberine chloride (CPU 86017) was first synthesized by the New Drug Center of China Pharmaceutical University and provided by Shan-dong Xinhua Pharmaceutical Industry Co. The 5 groups were the normal group (Ctl), PAH untreated group (PAH), and the 3 treatment groups (CPU 86017 20, 40, and 80 mg·kg-1·d-1,po, daily for 28 d from the day of MCT, sc). CPU 86017 was suspended freshly in 0.5% CMC-Na of 0.4%, 0.8%, and 1.6% (20, 40, and 80 mg·kg-1·d-1, sc). The normal and PAH groups were given an equal volume of CMC-Na.
The investigation was performed in accordance with the Guide for the Care and Use of Laboratory Animals in Jiangsu Province.
Hemodynamic analysis and heart weight index After 4 weeks of MCT medication and interventions with CPU 86017, rats were killed with urethane (1.5 mg/kg, ip). RVSP (right ventricular systolic pressure), CVP (central venous pressure), and blood pressure were measured using 2 catheters separately, as described previously[9]. Animals were then exsanguinated, and the hearts and lungs were dissected and weighed. The right ventricular remodeling was assessed by measuring the RVWI (right ventricle weight index): the RV weight to body weight ratio (RVW/BW, mg/g) and the RV-to-left ventricular (LV) plus septal (S) weight ratio [RVW/(LVW+SW), mg/g]. The improvement after drug intervention was calculated as follows: improvement % = (PAH-Treated)/(PAH-Normal)×100%.
ET-1, NO, NOS, superoxide dismutase (SOD), and malondialdehyde (MDA) assays Blood samples were collected in chilled vials containing 30 mL EDTA2Na and 20 mL Aprotinin. ET-1, NOS activity, SOD, and MDA in serum and lung tissue were measured using radioimmunoassay or the kits supplied by Nanjing Jiancheng Biotechnological Co, following the practice described previously[9].
Assessment of pulmonary artery remodeling Transverse sections of lungs, 4 µm in thickness, were cut at the right middle zones and stained with hematoxylin-eosin, then the wall thickness of small pulmonary arteries (diameter<150 µm) was analyzed using a colorful picture analysis system (Quantment 500), as described in a previous report[9]. The thickness (µm) and WT (wall thickness ratio) percentage value were determined as the average data of 10 fields per slice and the WT was calculated as below: WT%=(diameterext-diameterint)/diameterext×100.
mRNA expressions of preproET-1, iNOS, and eNOS in lung tissue Lung tissue was homogenized in TRIZOL, and then extracted with hydroxybenzene-chloroform. Reverse transcription-polymerase chain reaction (RT-PCR) was performed in 25 µL final volume under the following conditions: initial denaturalization at 94 oC for 5 min, denaturalization at 94 oC for 40 s, annealing for 40 s at 64 oC for preproET-1, at 58 oC for eNOS, at 58 oC for iNOS, and at 60 oC for β-actin, all extension at 72 oC for 1 min. The circular numbers of β-actin, preproET-1, eNOS, and iNOS were 25, 28, 26 and 28, respectively. The sequences of primers are shown in Table 1 and the procedures were the same as described previously[9]. The PCR products were electrophoresed and determined semi-quantitatively.

Full table
Functional assessment of isolated pulmonary arterial rings The pulmonary arteries were carefully isolated and cleaned of outer connective tissue in cold K-H solution saturated with 100% oxygen. The rings from each pulmonary artery (approximately 3 mm in length) were mounted vertically between 2 hooks in organ chamber myographs, which were filled with K-H solution and kept at 37 oC. Isometric tension was measured with force transducers (MPA-V). Constrictions of the isolated pulmonary arterial rings by KCl 100 mmol/L or phenylnephrine 1 µmol/L were performed in calcium free KH solution and after adding up to 2.5 mmol/L Ca2+ in the presence of either KCl or phenylephrine, separately[10–12].
Statistical analysis Data are presented as mean±SEM. For statistical evaluation one-way analysis of variance was used, following Dennett’s test. The Student Newman Keuls test was performed when the variance was equal, and the Games-Howell test was performed when variance was not equal. Pearson correlation analysis was also carried out on some indices. A value of P<0.05 was considered statistically significant.
Results
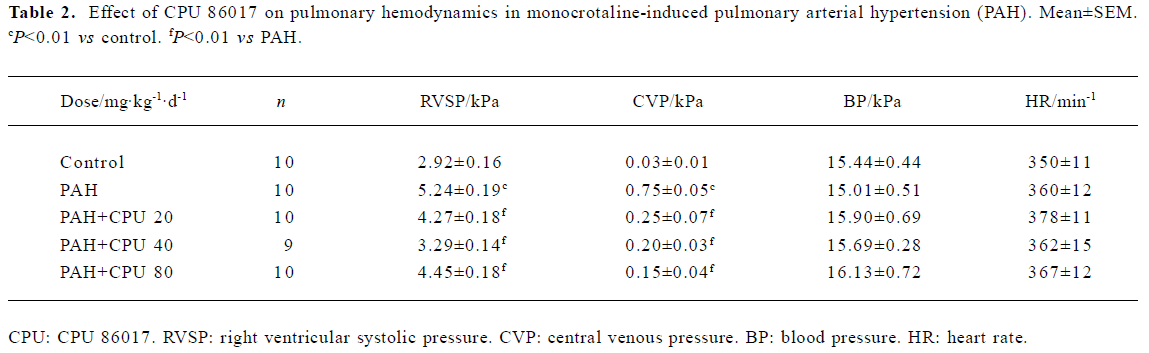
Hemodynamic changes The RVSP and CVP in MCT PAH group were elevated markedly by 79% (P<0.01) and 2400% (P<0.01) compared with normal, respectively. CPU 86017 20, 40, and 80 mg·kg-1·d-1 po decreased the RVSP by -18.5%, -37.2%, and -15.1%, compared to the PAH (P<0.01), respectively, and the reduction in CVP was by -66.7%, -73.3%, and -80.0%, respectively (P<0.01, Table 2).
Full table
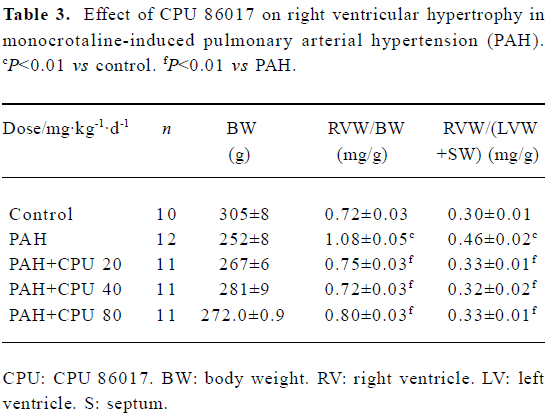
Regression in RVWI The RVWI in the control group was increased by 50.0% in RVW/BW and 53.3% in RVW/(RVW+SW) (P<0.01), respectively. Reduction in RVW/BW by CPU 86017 20, 40, and 80 mg·kg-1·d-1, po was -30.5%, -33.3%, and -25.9%, respectively, compared to the untreated group and in RVW/(RVW+SW) was -28.2%, -30.4%, and -28.2% (P<0.01), respectively. The remodeling of the right ventricle was regressed remarkably in each intervention group (Table 3).
Full table
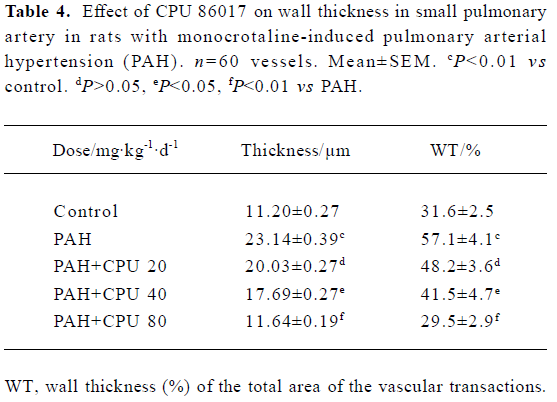
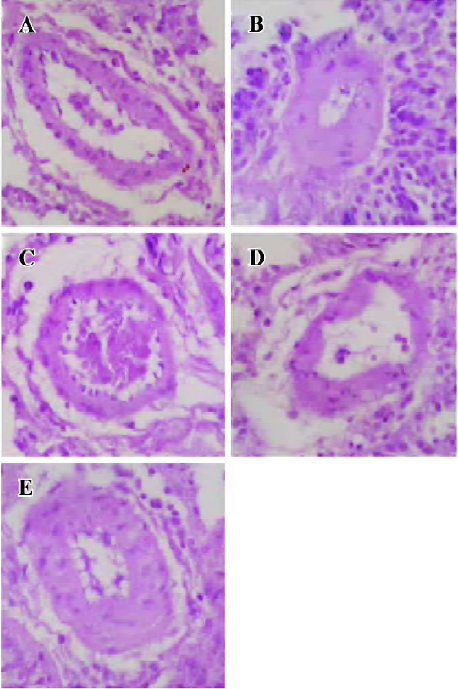
Regression in the vasculature of the pulmonary arteriole In PAH group, the thickness (μm) and the relative value of WT% (wall thickness percentage) of small pulmonary arteries (<150 µm in diameter) were increased by 106.0% and 80.7% (P<0.01) compared with the control group. In CPU 86017 80, 40, and 20 mg·kg-1·d-1 groups, thickness (μm) and the relative value of WT percentage of small pulmonary arteries decreased significantly compared with the untreated group (Table 4, Figure 2).
Full table
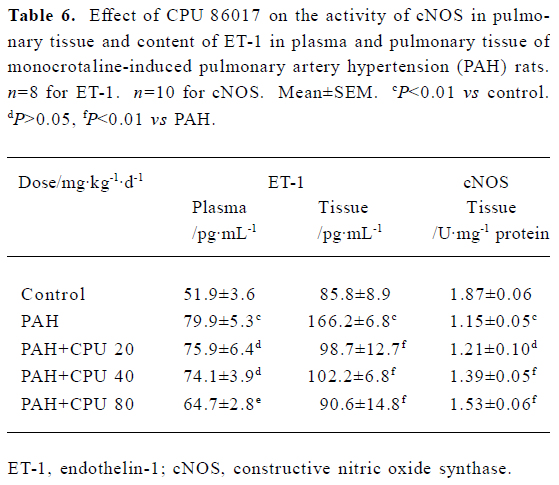
NO, ET-1, cNOS, SOD, and MDA in serum and lung tissue In the PAH group, the ET-1 in the serum and lungs was dramatically increased by 53.9% (P<0.01) and 93.7% (P<0.01), respectively. CPU 86017 decreased the content of ET-1 to the normal level in lung tissue, but was less effective in serum. The NO content was increased by 60.0% (P<0.01) in serum, but decreased by -52.5% (P<0.01) in pulmonary tissue, compared to that of the normal group. The level of NO was significantly increased in CPU 86017 80 and 40 mg·kg-1·d-1 groups in tissue, whereas the difference was not significant in serum. Activity of cNOS in tissue decreased by -38.5% (P<0.01) in the PAH group compared to the normal. The activity of cNOS was increased by 33.0% (P<0.01) in CPU 86017 80 mg·kg-1·d-1 po group compared to the PAH (Table 5, 6). The MDA was significantly increased in both the serum and lungs, whereas the activity of SOD decreased markedly. A significant reduction in MDA production was observed in all three CPU 86017 groups, which was associated with an increase in the SOD activity in the serum and lungs.

Full table
Full table
PreproET-1, iNOS, and eNOS mRNA expression Twenty-eight days after MCT injection, the mRNA expression of preproET-1, eNOS, and iNOS in the PAH group was upregulated by approximately 4, 6.5, and 2.7 times, respec-tively, against the normal lung tissue. The upregulation of eNOS and iNOS mRNA levels was significantly reversed in three CPU 86017 groups, and preproET-1 mRNA abundance was also reduced notably in CPU 86017 80 mg·kg-1·d-1 group versus the PAH group (Figure 3).

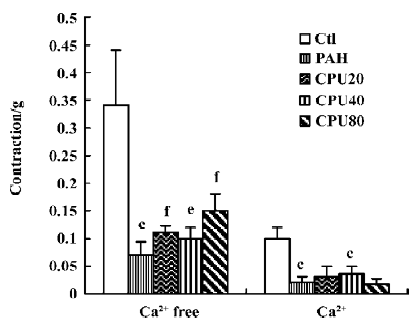
Isolated pulmonary artery activity Two sorts of pulmonary vascular activities were assessed using high KCl and phenylephrine in separate experiments. Each was evaluated in 2 phases of constriction of the isolated pulmonary artery rings in calcium-free medium, and up to 2.5 mmol/L calcium was added to the calcium-free medium in the presence of either high KCl or phenylephrine. The KCl induced vasoconstrictions in the calcium-free medium decreased markedly in the PAH group and recovered partially after CPU 86017 intervention. The constrictions by the calcium influx on adding calcium into calcium-free medium in the presence of KCl 100 mmol/L were impaired markedly in the untreated group, whereas there was no improvement in the CPU 86017-treated groups (Figure 4). The contractions by phenylephrine in the calcium-free medium were notable in the untreated group, and no improvement could be found after CPU 86017 treatments. A markedly impaired vascular tone of contractions by calcium influx resulting from an addition of Ca2+ into the medium in the presence of phenylephrine 1 μmol/L was found and vascular tension completely returned to normal in the CPU 86017 groups of 80 and 40 mg·kg-1·d-1, po (Figure 5).

Discussion
CPU 86017 is derived from chemical modification of the moiety of berberine and possesses a blocking effect on the K+ and Ca2+ channels[13], together with an improvement in the water solubility and availability in pharmacokinetics. The chemical reformation leads to a reinforcement of calcium antagonism, providing a potential beneficial use in the treatment of cardiovascular disorders[12].
The chronic inflammation of the lung tissue by MCT contributes to an insult to vascular endothelium and myocardium, consequently resulting in a substantial increase in ET-1 release, which promotes the pathological progression of PAH. The voltage-dependent calcium channels are coupled to ETA receptors by G proteins; therefore, an enhancement of the calcium influx is anticipated to follow an excess ET-1 release, as we found in the PAH. The activation of the L-type Ca2+ channels caused by an excess ET-1 in the myocardium and vasculature is an important etiological factor resulting in maladjustment and remodeling changes in the pulmonary vessels and the right ventricle.
The ET-1 closely linked with the ROS system is an important pathway involved in cardiovascular derangement[14]. One of the up-stream events in the pathway to the tissue ET-1 is the mRNA of preproET-1, which is also elevated significantly. An excess of ET-1 binding to the ETA receptors stimulates the protein kinase C (PKC), mitogen-activated protein kinase (MAPK) pathway, finally leading to an abnormality of phenotype of genes encoding the preproET-1, iNOS, and cNOS, matrix formation, as well as to pulmonary VSMC proliferation[15–17].
A low level of NO and elevated ET-1 in the pulmonary tissue is a result of endothelial damage and dysfunction by MCT. MCT selectively damages pulmonary endothelium, reducing NO production and secretion. The vascular NO is biosynthesized and released in a constant manner, which is necessary to maintain the normal vascular vasodilatation response. The bioavailability of NO (ie basal release) is compromised significantly, resulting in impaired vascular activity in the PAH group. An abnormally low NO exerts a suppressive effect on the proliferation of smooth muscle cells, because of an excess of ET-1, which mediates an activation of MAPK pathway. An increased serum NO in the PAH group is a result of activated activity of the iNOS resulting from the stimulation of chronic inflammation by MCT, which accelerates both pulmonary artery hypertension and portal hypertension[18].
The ROS play an important role in remodeling of the small pulmonary artery, as they proliferate smooth muscle cells by activating the phosphorylation of ERK/MAPK. The ET-1 stimulates the downstream events in the pathway; that is, activation of iNOS and ROS. It is likely that a circulated activation mechanism takes place between the upstream and the downstream to exacerbate an activation of the ET pathway involved in the pathological process. An increment of ROS causes an increase in the abundance of mRNA of prepro-ET-1, eNOS, and iNOS, attributable to the enhancement of the transcription process in the nucleus[19,20]. This is easily reversed by suppression of the ET–ROS pathway after treatment with CPU 86017.
An intervention to interrupt the targets located at either the upstream or the downstream of the ET pathway is capable of relieving the disturbance in the PAH by MCT. The insult to the pulmonary endothelium and the later developed myocardial insufficiency of the right ventricle are the main factors contributing to excess ET-1 and the over-stimulation of the ET pathway. The overactivated ET pathway can be reversed indirectly by the calcium antagonism and antioxida-tive effects of CPU 86017. Actually an attenuation of the activated ET pathway by CPU 86017 in the MCT PAH in rats can not be ascribed to a direct blocking action on ET receptors, but ascribed to a calcium antagonism relieving an injury of the pulmonary endothelium and the myocardium and rebalancing an interruption of the ROS formation. A reduced formation of the ET-1 and an indirect blockade of endothelin receptors to alleviate its downstream events are achieved using CPU 86017 chronic medication.
Plasma ET-1 was less responsive than the pulmonary origin ET-1to CPU 86017 treatment. The ET can be sourced from the two: physiological and pathological conditionas, sush as the ET in the affected lung is totally from the insult from MCT. In the blood stream the majority of ET is released from the organ under normal conditions such as the central nervous system, the kidney, endometrium, and normal tissue[21], but ET from the affected lung contributes to a small portion of plasma ET-1. So only a heavy inhibition of the release from the pathological lung by the high dose of CPU 86017 could provide a reduction in the seral total ET, but not the middle and small doses. In contrast, paracrine and autocrine ET-1 is effective in the local region. The pulmonary origin of ET-1 was the main pathological factor and responded well to the CPU 86017 treatment.
We found that eNOS mRNA expression was significantly increased in MCT-induced PAH, and was up to 6.5 times higher than that in normal group, whereas both NOS activity and NO content decreased in the same way as reported by Resta et al[22]. Such a contradiction might be contributed to damage to endothelium by MCT with which the eNOS can not be converted into a normally active form. The maladjustment resulted from phosphorylation of threonine[23], which causes defective activity of the eNOS, contributing to PAH progression[24].
In conclusion, an activated ET-ROS pathway damages the endothelium system which results in consequent pulmonary vasculature derangement in PAH by MCT in rats. By suppressing the ET pathway, CPU 86017 relieves MCT PAH.
References
- Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis 2002;45:173-202.
- Tuder RM, Cool CD, Yeager M, Taraseviciene-Stewart L, Bull TM, Voelkel NF. The pathobiology of pulmonary hypertension. Endothelium. Clin Chest Med 2001;22:405-18.
- Wort SJ, Woods M, Warner TD, Evans TW, Mitchell JA. Endogenously released endothelin-1 from human pulmonary artery smooth muscle promotes cellular proliferation: relevance to pathogenesis of pulmonary hypertension and vascular remodeling. Am J Resp Cell Mol Biol 2001;25:104-10.
- Michelakis ED. The role of the NO axis and its therapeutic implications in pulmonary arterial hypertension. Heart Fail Rev 2003;8:5-21.
- Alexandru B, Bogdan MA. Monocrotaline induces pulmonary hypertension in animal models. Pneumologia 2001;50:85-9.
- Wang HL, Li SB, Dai DZ. Effects of CPU 86017 on L-type calcium current in single guinea pig hypertrophic ventricular myocytes. J China Pharm Univ 2004;35:259-62.
- Hao JM, Dai DZ, Yu F. The oxidative stress is enhanced after L-thyroxin medication and suppressed by CPU 86017. Prog Pharm Sci 2005;29:417-21.
- Wang YQ, Shi YP, Dai DZ. The therapeutic effects of CPU 86017 on acute and chronic congestive cardiac failure mediated by reducing ET-1, NOS and oxidative stress in rats. Drug Dev Res 2004;63:22-32.
- Zhang TT, Dai DZ. Therapeutic effects of nifedipine on pulmonary hypertension and mechanism involved. Prog Pharm Sci 2003;27:227-9.
- Dai DZ, Feng Y, Li HT. Blockade on sodium, potassium, and calcium channels by a new antiarrhythmic agent CPU 86017. Drug Dev Res 1996;39:138-46.
- Dai DZ, Jiang JM. p-Chlorobenzyltetrahydroberberine inhibits vascular smooth muscle contractions caused by Ca2+. Acta Pharmacol Sin 1998;19:543-7.
- Dai DZ, He Y, Huang FH. Comparison of inhibitory activity of berberine derivate CPU 86017 and berberine on vascular contraction. J China Pharm Univ 2000;31:447-50.
- Dai DZ, Hu HJ, Zhao J, Hao XM, Yang DM, Zhou PA, et al. Blockade of L-type calcium channel in myocardium and calcium-induced contractions of vascular smooth muscle by CPU 86017. Acta Pharmacol Sin 2004;25:416-23.
- Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004;110:1269-75.
- Mazzocchi G, Rossi GP, Malendowicz LK, Champion HC, Nussdorfer GG. Endothelin-1 [1-31], acting as an ETA-receptor selective agonist, stimulates proliferation of cultured rat zona glomerulosa cells. FEBS Lett 2000;487:194-8.
- Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M, Karliner JS. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol 2000;32:565-76.
- Herman WH, Simonson MS. Nuclear signaling by endothelin-1: a Ras pathway for the activation of the c-fos serum response element. J Biol Chem 1995;270:654-61.
- Perazzo J, Eizayaga F, Romay S, Bengochea L, Pavese A, Lemberg A. An experimental model of liver damage and portal hypertension induced by a single dose of monocrotaline. Hepatogastro-enterology 1999;46:432-5.
- Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, et al. Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J Biol Chem 2005;280:17497-506.
- Rodriguez JA, Nespereira B, Perez-Ilzarbe M, Eguinoa E, Paramo JA. Vitamins C and E prevent endothelial VEGF and VEGFR-2 overexpression induced by porcine hypercholesterolemic LDL. Cardiovasc Res 2005;65:665-73.
- Hsu YH, Huang SC. Immunohistochemical localization of endothelin-converting enzyme-1 in neuroendocrine tumors and normal human tissue. Kaohsiung J Med Sci 2003;19:555-62.
- Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol 1999;276:H699-H708.
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 2001;88:E68-E75.
- Murata T, Sato K, Hori M, Ozaki H, Karaki H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem 2002;277:44085-92.