
N-Acetyl-L-cysteine and pyrrolidine dithiocarbamate inhibited nuclear factor-κB activation in alveolar macrophages by different mechanisms
Introduction
Nuclear factor kappa B (NF-κB) is a collective name for inducible dimeric transcription factors comprising of members of the Rel family. Eight members of the Rel protein family including c-Rel, NF-κB1 (p50/p105), NF-κB2 (p52/p100), Rel A (p65), Rel B, the Drosophila proteins Dorsal, Dif, and Relish have been cloned and characterized[1], and all share a conserved Rel homology domain (RHD) that is responsible for dimerization, nuclear localization, and DNA binding/transcription activation. NF-κB is found in essentially all cell types and it helps to govern the expression of genes involved in such disparate processes as growth, development, inflammatory/immune response, auto-regulation, and transcription of viral genomes[2]. Some studies have also shown that NF-κB plays an important role in the expression of matrix metalloproteinases (MMP)[3,4]. A family of structurally related proteins known as the inhibitors of NF-κB (IκB) are responsible for the regulation of the DNA binding activity and nucleo/cytoplasmic distribution of NF-κB. All IκB share an ankyrin repeat domain (ARD) that interfaces with the RHD of NF-κB homo/heterodimers, forming a stable inhibited complex. IκBα binds the p65/p50 heterodimer (the most ubiquitous and biologically active NF-κB dimmer) and p65/c-Rel heterodimers, and is the best characterized protein in this family. The association IκBα with NF-κB disrupts DNA binding and masks the nuclear localization signals (NLSs) located in the C-terminal region of the RHD, which is believed to impede nuclear translocation of NF-κB[5]. So NF-κB is normally sequestered in the cytoplasm of non-stimulated cells and consequently must be translocated into the nucleus to function. A variety of extracellular stimuli including in tumor necrosis factor (TNF)-α and interleukin (IL)-1 cause the rapid phosphorylation, ubiquitination, and ultimately proteolytic degradation of IκBα, which allows NF-κB to translocate to the nucleus where it regulates gene transcription[6,7]. The multisubunit IκB kinase (IKK) responsible for inducible IκBα phosphorylation is the point of convergence for most NF-κB-activating stimuli. The IKK complex consists of 2 catalytic subunits, IKKα and IKKβ, and a scaffold molecule, the NF-κB essential modulator (or IKKβ). Gene depletion studies have demonstrated that IKKβ, but not IKKα, plays an essential role in NF-κB activation mediated by LPS, TNF-α, and IL-1[8–10].
MMP that can degrade most components of the extracellular matrix (ECM) play an important role in the pathogenesis of chronic obstructive pulmonary disease (COPD)[11,12]. Ya-qing LI, et al (unpublished data), have shown that N-acetyl-L-cysteine (NAC) and pyrrolidine dithiocarbamate (PDTC) can down-regulate the expression of MMP-9 induced by TNF-α in alveolar macrophages (AM) from patients with COPD via a NF-κB-mediated signal pathway. Because NAC and PDTC are antioxidants, and both can inhibit the activation of NF-κB, it has been postulated that reactive oxygen species (ROS) may act as second messengers leading to NF-κB activation[13]. However, this hypothesis does not explain why other antioxidants, such as epigallocatechin-gallate (EGCG) and trolox, failed to inhibit NF-κB activation induced by TNF-α[14]. Therefore it was necessary to study the pharmacological mechanisms of NAC and PDTC as inhibitors of NF-κB-mediated pathways. So in the present study, their effects on the phosphorylation of IKKβ, IKKα, and IκBα were investigated, and NF-κB activity induced by TNF-α and IL-1 was also explored.
Materials and methods
Experimental subjects Twenty patients with COPD were recruited from Tongji Hospital, Wuhan, China. The diagnosis of COPD was made on the basis of medical history, clinical symptoms and spirometric data, according to the criteria of the Global Initiative for Chronic Obstructive Lung Disease[15]. Pulmonary function tests were performed to determine forced vital capacity (FVC) and forced expiratory volume in one second (FEV1). Chronic airflow obstruction was defined as having an FEV1/FVC ratio of <70% and a postbronchodilator FEV1 value that was <80% of the predicted value. Subjects were excluded if they had asthma, recent exacerbation of COPD, or had received recent treatment with oral corticosteroids. All patients gave written informed consent before participating in the study, which was approved by the Ethics Committee of Tongji Hospital.
Bronchoalveolar lavage and AM culture Bronchoscopy and bronchoalveolar lavage (BAL) were performed as described previously[16]. Three 30 mL aliquots of sterile saline solution were instilled using a bronchoscope to the subseg-mental bronchi of the middle lobe. The recovered fluid was strained through surgical gauze to remove debris and mucus, and was then centrifuged (400×g for 10 min). BAL cells were washed twice in phosphate-buffered saline (PBS), and resuspended in RPMI-1640 medium (Hyclone, Logan, Utah, USA) containing 10% fetal calf serum (FCS, Gibco, Grand Island, New York, USA). The cells were allowed to adhere for 2.5 h at 37 °C in a humidified 5% CO2 incubator. Nonad-herent cells were removed by washing twice with PBS. Adherent cells were identified by Giemsa staining. Only AM and bronchial epithelial cells adhered to cell culture plates; neutrophils did not adhere and bronchial epithelial cells constituted less than 1% of adherent cells. AM viability was measured by trypan blue exclusion.
Cell treatments AM that had been obtained from BAL fluid (BALF) were seeded into 24-well cell culture plates at a concentration of 1×109 cells/L. First the cells were incubated for 1.5 h with either NAC (Sigma, St Louis, Missouri, USA) or PDTC (Sigma, USA), and they were then stimulated by either TNF-α (Biosea, Beijing, China) or IL-1 (Biosea, China). The AM and supernatant were harvested, and the supernatant was stored at -80 °C until use.
Western blotting Cytoplasmic proteins were extracted from the cultured AM with NE-PER Nuclear and Cytoplasmic Extraction Reagent (Pierce, Woburn, Massachusetts, USA) according to the manufacturer’s instructions. The samples containing 20 µg protein were incubated for 5 min in boiling water. Prestained protein molecular weight markers (Fermentas, Burlington, Ontario, Canada) were included in each gel. Proteins were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then electroblotted onto Hybond-ECL nitrocellulose membrane. Non-specific binding sites were blocked by immersing the membranes in Tris-buffered saline Tween (TBS-T) for 1 h at room temperature on an orbital shaker. Membranes were then incubated in primary antibodies for 1 h at room temperature on an orbital shaker. The membranes were then washed 5 times for 5 min each in TBS-T and incubated in the diluted peroxidase-conjugated immunopure goat anti-rabbit IgG (H+L) (Pierce, USA) for 1 h at room temperature on an orbital shaker. After the membranes were again washed 5 times for 5 min each in TBS-T, the proteins were visualized using enhanced chemiluminescence solution (ECL). The images were developed on X-ray film and the band densities were analyzed by using a UVP-GDS8000 gel analysis system. As a loading control, the same membranes were stripped and blotted with anti-α-tubulin antibodies. The primary antibodies were obtained from the following sources: phospho-IκBα (Ser32) antibody, phospho-IKKalpha (Ser180)/IKKbeta (Ser181) antibody from Cell Signaling Technology (Danvers, Massachusetts, USA); rabbit monoclonal antibody against human α-tubulin from Santa Cruz Biotechnology (Santa Cruz, California, USA).
Electrophoretic mobility shift assay Nuclear protein was extracted from the cultured AM with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, USA) according to the manufacturer’s instructions. The sequences of biotin-NF-κB DNA probes that were double-strand end-labeled by biotin were 5'-xGCCTGGGAAAGTCCCCTCAACT-3' (sense) and 5'-xAGTTGAGGGGACTTTCCCAGGC-3' (anti-sense) (biotins are represented by x). The sequences of Unlabeled-NF-κB DNA probes were 5'-GCCTGGGAAAGT-CCCCTCAACT-3' (sense) and 5'-AGTTGAGGGGACTTT-CCCAGGC-3' (antisense) (SBS Biotechnology, Shanghai, China). NF-κB DNA binding activity was detected by using a LightShift Chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Pierce, USA) according to the manufac-turer’s instructions as follows. A 5% native polyacrylamide gel was prepared in 0.5×TBE (Tris-Borate EDTA), and pre-electrophoresed for 1 h. at 100 V in 0.5×TBE. Binding reaction mixture containing 50 ng/µl Poly(dI·dC), 0.05% NP-40, 50 mmol/L KCl, 5 mmol/L MgCl2 and 10 mmol/L ethylenediamine tetraacetic acid (EDTA) were incubated at room temperature for 20 min. Five microliters of 5× loading buffer was added to each 20 µL of binding reaction mixture, mixed, and 20 µL of each sample was loaded onto the polyacrylamide gel. Samples were electrophoresed at 100 V until the bromophenol blue dye had migrated approximately two-thirds to three-quarters of the length of the gel, and then the binding reactions were electroblotted onto nylon membrane at 380 mA (~100 V) for 1 h. The membrane was cross-linked for 10 min at a distance of approximately 0.5 cm from an ultraviolet lamp equipped with 254 nm bulbs, and biotin-labeled DNA was visualized by using ECL. The images were developed on X-ray film and the band densities were analyzed by using a UVP-GDS8000 gel analysis system (Ultra-Violet Products Ltd, Cambridge, UK).
Statistical analysis Data were analyzed with SPSS for Windows Version 11.5. All results are expressed as mean±SD. Statistical evaluations of the data were performed with one-way analysis of variance (ANOV) and the paired-samples t-test, and multiple comparisons in ANOV were analyzed by using the Student-Newman-Keuls test. For all tests, P<0.05 was considered to be statistically significant.
Results
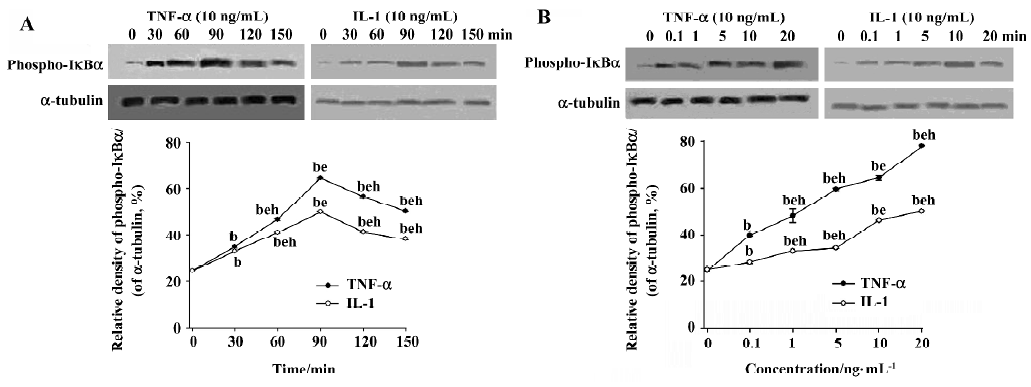
Effects of TNF-α and IL-1 on phosphorylation of IκBα in COPD AM After stimulation with TNF-α (10 ng/mL) or IL-1 (10 ng/mL), cytoplasmic proteins of the cultured AM were collected at various time points (0 min, 30 min, 60 min, 90 min, 120 min and 150 min). The phosphorylation level of IκBα increased in a time-dependent manner, and peak induction of the phosphorylation was observed at 90 min (Figure 1A). To determine dose responsiveness, the cultured AM were incubated for 90 min with TNF-α and IL-1 at concentrations from 0.1 ng/mL to 20 ng/mL. Both TNF-α and IL-1 can induce the phosphorylation of IκBα in a dose-dependent manner (P<0.05; Figure 1B).
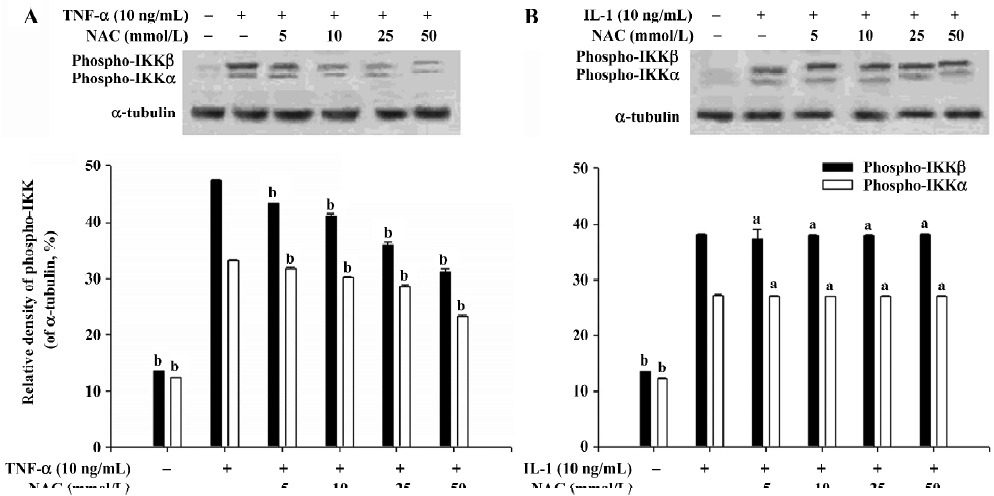
Effect of NAC on phosphorylation of IKKα and IKKβ When AM were stimulated by TNF-α or IL-1 at a concentration of 10 ng/mL, the levels of both phospho-IKKβ and phospho-IKKα were significantly elevated (P<0.05; Figure 2). Our results showed that NAC had no effect on the phosphorylation of IKKβ and IKKα induced by IL-1 (Figure 2B). However, the phosphorylation of IKKβ and IKKα induced by TNF-α in the AM pretreated with NAC was significantly inhibited (P<0.05; Figure 2A). The results suggest that the processes upstream of IKK activation induced by TNF-α can be inhibited by NAC, which indirectly results in inhibition of the phosphorylation of IKKβ and IKKα.
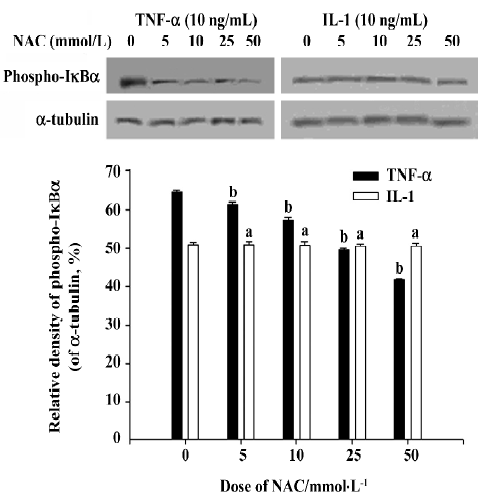
Effects of NAC on phosphorylation of IκBα induced by TNF-α and IL-1 NAC could inhibit the phosphorylation of IκBα induced by TNF-α (Figure 3), but not affect the phosphorylation of IκBα induced by IL-1 (Figure 3). Similarly, EMSA showed that the activation of NF-κB induced by TNF-α was inhibited by NAC in a dose-dependent manner (Figure 4A), but that NAC did not affect the activation of NF-κB induced by IL-1 (Figure 4B). However, if 50 mmol/L NAC was directly added into the nuclear extracts, NF-κB DNA binding activity was not directly inhibited by it in vitro (Figure 5).
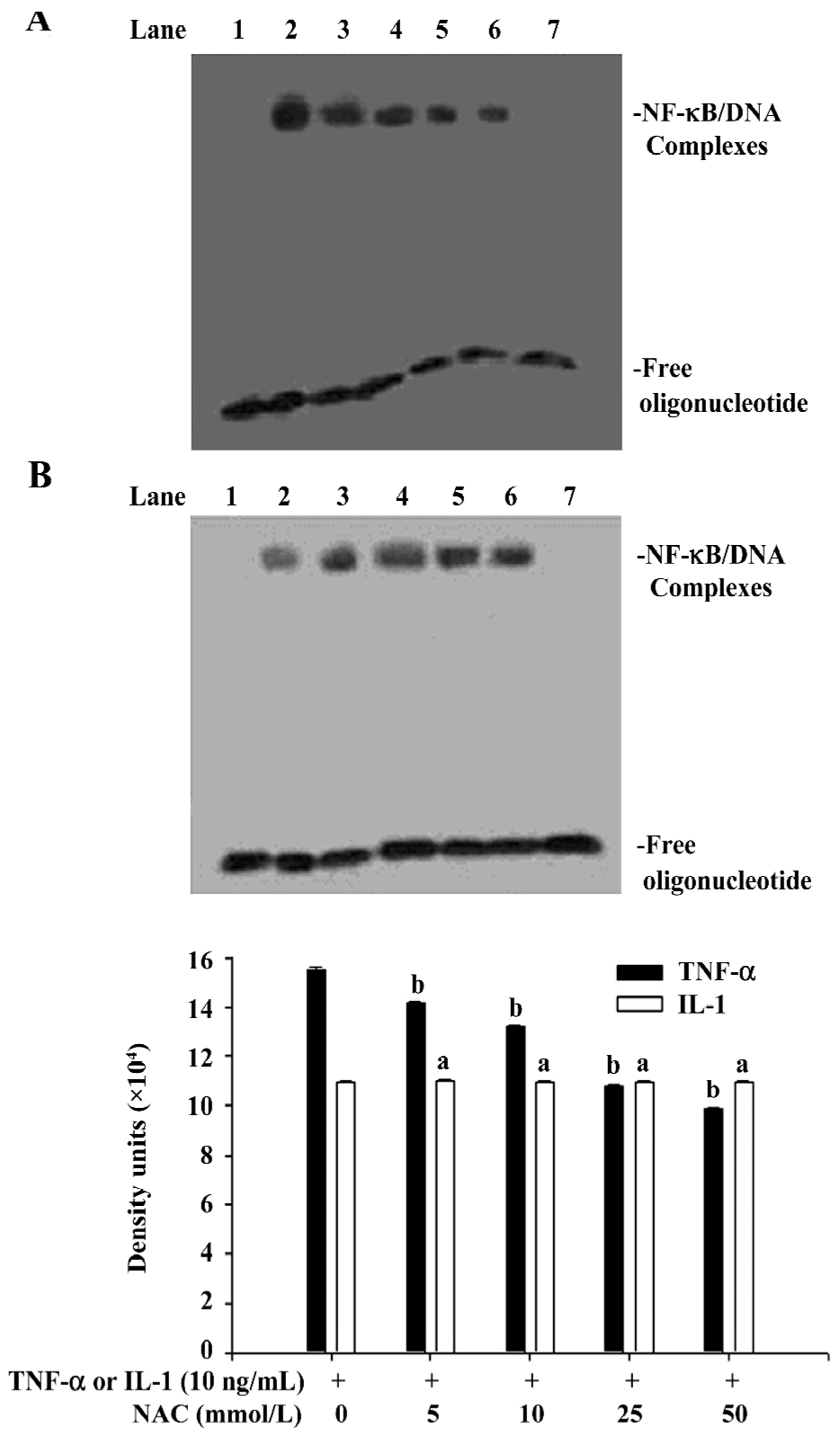
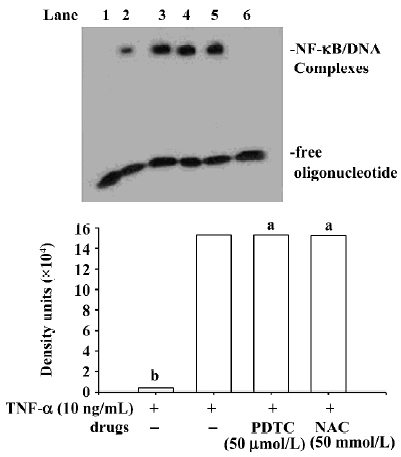
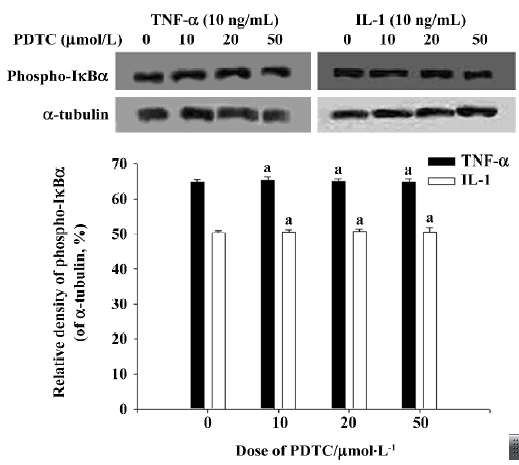
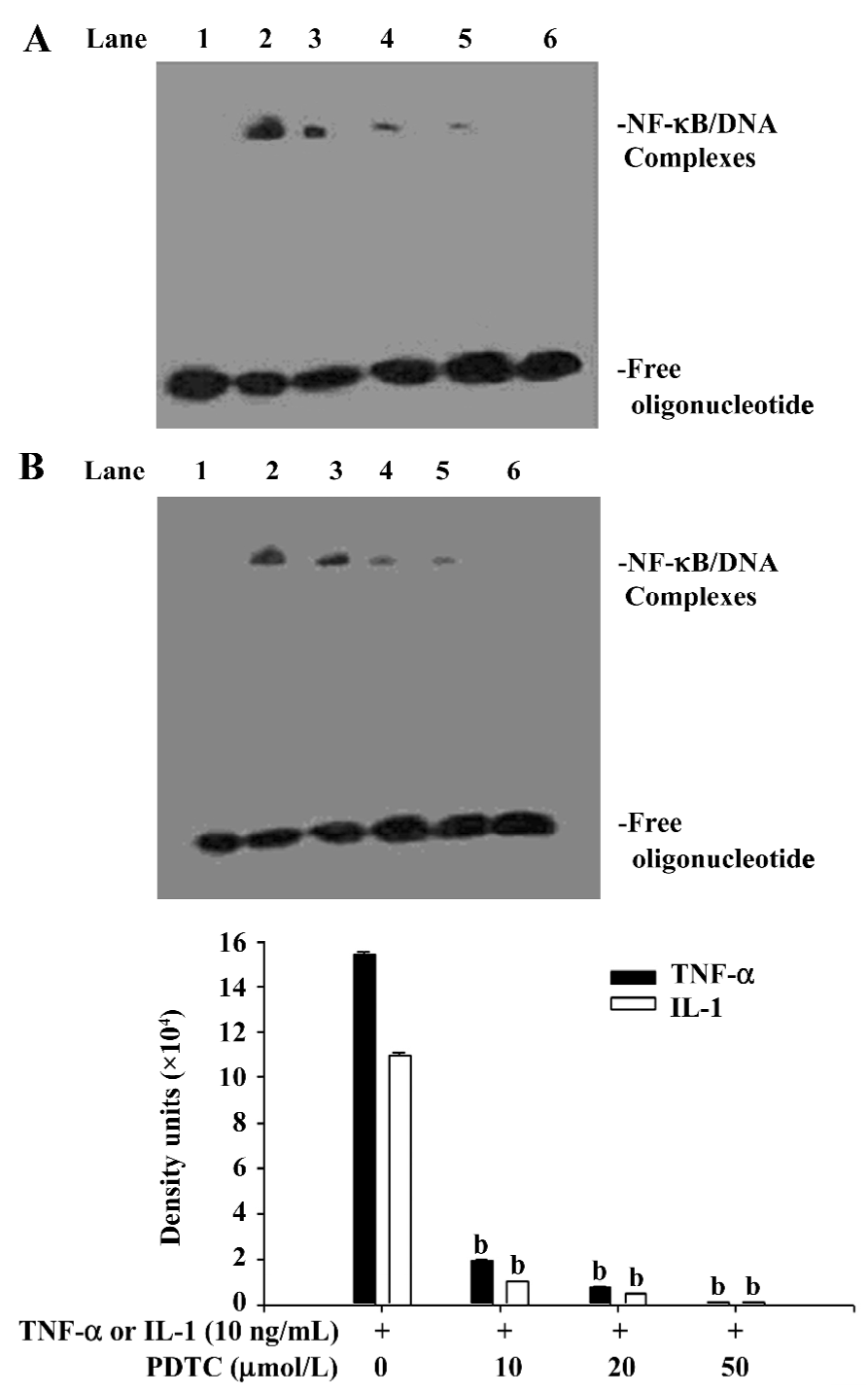
Effect of PDTC on the release of IκB AM were pretreated for 1.5 h with PDTC at concentrations ranging from 10 µmol/L to 50 µmol/L, and then were stimulated for 90 min by TNF-α or IL-1 at a concentration of 10 ng/mL. Cytoplasmic and nuclear extracts were also prepared. Western blot analysis showed that PDTC does not inhibit the phosphorylation of IκBα induced by TNF-α and IL-1 (Figure 6), whereas EMSA showed that the activation of NF-κB induced by both TNF-α and IL-1 can be significantly inhibited by PDTC in a dose-dependent manner (P<0.05; Figure 7). However, PDTC (50 µmol/L) directly added into the nuclear extracts, had no direct effect on NF-κB DNA binding activity in vitro (Figure 5).
Discussion
In the classical NF-κB signaling pathway, upon stimulation by TNF-α or IL-1, the IKK signalosome composed of IKKα, IKKβ, and IKKγ is activated, which leads to the phosphorylation of IκB on 2 conserved N-terminal serine residues. Then phosphorylated IκB are ubiquitinated and subsequently degraded by the S26 proteasome. So NF-κB is activated, and then translocated into the nucleus. Finally, it binds to a decameric consensus motif in target genes, resulting in their transcription[6,17]. In the present study we found that the phosphorylation of IKKα, IKKβ, and IκBα induced by TNF-α can be inhibited by NAC. But we also found that NAC had no effect on the phosphorylation of IKKα, IKKβ, and IκBα induced by IL-1. Although the IKK responsible for IκBα phosphorylation is the point of convergence for the activation of NF-κB induced by either TNF-α or IL-1, there are different upstream mechanisms that result in IKK activation. Some studies have indicated that activation of TNF receptor (TNFR) 1 triggered by TNF-α can result in strong activation of IκB kinase (IKK) via a pathway including TNFR1-associated protein with death domain (TRADD), TNFR-associated Factor (TRAF) 2, receptor-interacting protein (RIP) and MAP kinase ERK kinase kinase 3 (MEKK3) [18–20]. Furthermore, upstream of IL-1-triggered IKK activation, Toll/interleukin-1 receptor domain-containing adapter protein (TIRP), interleukin-1 receptor associated kinase 1 (IRAK1), IRAK4 and TRAF6 played important roles[21,22]. So although NAC does not directly inhibit the phosphorylation of IKKβ and IKKα, it does inhibit the processes upstream of IKK activation induced by TNF-α. The present study showed that NAC could inhibit the activation of NF-κB induced by TNF-α, but not interfere with the activation of NF-κB induced by IL-1. EMSA also showed that NAC didn’t directly inhibit NF-κB DNA binding activity in vitro. Therefore, we think that NAC can inhibit the upstream of IKK activation induced by TNF-α, which blocked TNF-α-stimulated NF-κB activation. We know that the extracellular domain of TNFR is composed of 4 conserved cysteine-rich repeats, and 4–6 cysteines were involved in disulfide bridges in the repeats [23]. Because NAC can cause structural changes in TNF receptors by reducing the disulfide bridges, it can lower the affinity of TNFR1 or TRAF2, which is perhaps related to the action of NAC.
PDTC is a fairly specific inhibitor of NF-κB induction, which has no significant influence on the binding activity of oct-1 or proteins binding to a cAMP-response element and the GC-rich binding motif of Spl[24]. Moreover, PDTC also blocks NF-κB activation independently of the inducing agent and cell line[24]. However, the nature of the pharmacological mechanism by which it inhibits NF-κB activation remains controversial. In the present study, we found that PDTC had no effects on the phosphorylation of IκBα induced by TNF-α or IL-1, but that the activation of NF-κB induced by both TNF-α and IL-1 could be significantly inhibited by PDTC. We also found that PDTC does not directly inhibit NF-κB DNA binding activity in vitro, which accords with the findings of Schreck et al[24]. So it is evident that PDTC can prevent the degradation of IκBα, and prevent the release of IκB from NF-κB, which results in the inhibition of NF-κB activation. It has been shown that ubiquitin-protein ligases (E3) play a key role in the ubiquitin-mediated proteolytic cascade, conferring high specificity and selectivity on the system[25,26]. Specific ring finger complexes (SCF complexes) belong to the ring finger domain-containing E3 family, and SCFβ-TrCP (transducin repeat-containing protein) is one member of the SCF complex. When IκBα is phosphorylated on Ser32 and Ser36, it will be recognized by SCFβ-TrCP, then ubiquitinated, and finally degraded by the 26S proteasome[27–29]. So we speculate that PDTC can inhibit the activity of SCFβ-TrCP, which protects IκBα from being degraded via the ubiquitylation-proteasome proteolytic pathway. Although signals that induce the phosphorylation of IêB can also cause the phosphorylation of NF-κB proteins[30], recent evidence indicates that the phosphorylation of NF-κB subunit p65 can modulate NF-κB transcription activity but not affect nuclear translocation or DNA binding affinity[31,32]. This finding notwithstanding, it was useful to further study whether PDTC can directly inhibit the phosphorylation of NF-κB.
In short, although both NAC and PDTC are inhibitors of NF-κB activation, the sites they inhibit are different, and their function seems to have nothing to do with their antioxidant properties. These findings cast a new light on the treatment of COPD on the basis of this new property of NAC.
Acknowledgement
We thank Sheng-dao XIONG, Jian-ping ZHAO, and Jin LIU for their technical assistance.
References
- Baldwin AS Jr. The NF-kappaB and IkappaB proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649-81.
- Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998;16:225-60.
- Hozumi A, Nishimura Y, Nishiuma T, Kotani Y, Yokoyama M. Induction of MMP-9 in normal human bronchial epithelial cells by TNF-alpha via NF-kappaB-mediated pathway. Am J Physiol Lung Cell Mol Physiol 2001;281:L1444-52.
- Smola-Hess S, Schnitzler R, Hadaschik D, Smola H, Mauch C, Krieg T, et al. CD40L induces matrix-metalloproteinase-9 but not tissue inhibitor of metalloproteinases-1 in cervical carcinoma cells: imbalance between NF-κB and STAT3 activation. Exp Cell Res 2001;267:205-15.
- Johnson C, van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J 1999;18:6682-93.
- Ting AY, Endy D. Signal transduction. Decoding NF-kappaB signaling. Science 2002;298:1189-90.
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-kappaB activity. Annu Rev Immunol 2000;18:621-63.
- Yang F, Tang E, Guan K, Wang CY. IKKbeta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 2003;170:5630-5.
- Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev 2000;14:1729-33.
- Prajapati S, Gaynor RB. Regulation of IkappaB kinase (IKK)gamma/NEMO function by IKKbeta-mediated phosphorylation. J Biol Chem 2002;277:24331-9.
- Kang MJ, Oh YM, Lee JC, Kim DG, Park MJ, Lee MG, et al. Lung matrix metalloproteinase-9 correlates with cigarette smoking and obstruction of airflow. J Korean Med Sci 2003;18:821-27.
- Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 2005;54:31-6.
- Flohé L, Brigelius-Flohé R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med 1997;22:1115-26.
- Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, et al. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J 2003;22:3356-66.
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Seattle: GOLD; 2005. Available from URL: http://www.goldcopd.com/
- Montano M, Beccerril C, Ruiz V, Ramos C, Sansroes RH, Gonzalez-Avila G. Matrix metalloproteinases activity in COPD associated with wood smoke. Chest 2004;125:466-72.
- Ghosh S, Karin M. Missing pieces in the NF-κB puzzle. Cell 2002;109 Suppl:S81-S96.
- Wajant H, Grell M, Scheurich P. TNF receptor associated factors in cytokine signaling. Cytokine Growth Factor Rev 1999;10:15-6.
- Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal 2001;13:389-400.
- Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol 2001;2:620-4.
- Bin LH, Xu LG, Shu HB. TIRP, a novel toll/interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J Biol Chem 2003;278:24526-32.
- Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)-IRAK-tumor necrosis factor receptor-associated factor 6 (TRAF6) complex. J Biol Chem 2003;278:10952-56.
- Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 1995;5:392-9.
- Schreck R, Meier B, Mennel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor-kappaB activation in intact cells. J Exp Med 1992;175:1181-94.
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002;82:373-428.
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem 2001;70:503-33.
- Yaron A, Hatzubai A, Davis M, Lavon I, Manning AM, Andersen JS, et al. Identification of the receptor component of the IkappaB alpha-ubiquitin ligase. Nature 1998;396:590-4.
- Maniatis T. A ubiquitin ligase complex essential for the NF-kappaB, Wnt/Wingless, and Hedgehog signaling pathways. Genes Dev 1999;13:505-10.
- Sears C, Olesen J, Rubin DM, Finley D, Maniatis T. NF-kappaB p105 processing via the ubiquitin-proteasome pathway. J Biol Chem 1998;273:1409-19.
- Schmitz ML, dos Santos Silva MA, Baeuerle PA. Transactivation domain 2 (TA2) of p65 NF-kappa B. Similarity to TA1 and phorbol ester-stimulated activity and phosphorylation in intact cells. J Biol Chem 1995;270:15576-84.
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the IkappaB kinase alpha and beta subunits in liberating nuclear factor-kappaB (NF-kappaB) from IkappaB and in phosphorylating the p65 subunit of NF-kappaB. J Biol Chem 2002;277:3863-9.
- Wang D, Baldwin AS Jr. Activation of nuclear factor-kappaB-dependent transcription by tumor necrosis factor-alpha is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem 1998;273:29411-6.