
C333H, a novel PPARα/γ dual agonist, has beneficial effects on insulin resistance and lipid metabolism1
Introduction
The link between obesity and type 2 diabetes has been appreciated for a long time. Obesity has been demonstrated to be associated with lipid metabolic disorders, which commonly increase the risk of insulin resistance[1]. Insulin resistance can lead to several secondary complications, such as hypertension, atherosclerosis, and coronary artery disease. Type 2 diabetes is also characterized by insulin resistance, and in most cases hyperlipidemia[2]. Therefore, new therapies for adjusting insulin level and lipid metabolism are required to control type 2 diabetes.
Peroxisome proliferator-activated receptors (PPAR) are ligand-activated transcription factors that belong to the nuclear receptor superfamily[3,4]. The PPAR have specific tissue distributions and play a pivotal role in regulating the expression of a large number of genes involved in glucose and lipid metabolism[5]. It has been shown that PPARα is expressed at high levels in the liver, and mainly regulates lipid metabolism. Hypolipidemic fibrate-class drugs effectively activate PPARα. In contrast, PPARγ is mainly distributed in adipose tissue and skeletal muscle, and regulates glucose metabolism. The thiazolidinediones (TZD), which are antidiabetic drugs, are PPARγ agonists.
Currently, most agonists that are clinically used against PPAR specifically activate either PPARα or PPARγ. As a high-affinity PPARγ agonist, rosiglitazone has been demonstrated to have a variety of clinical effects, including improving insulin sensitivity and glucose tolerance[6,7]; however, its activity with respect to PPARα is weak, so its ability to regulate lipids is limited. In contrast, PPARα agonists such as fibrate and fenofibrate primarily decrease serum triglyceride levels and increase high density lipoprotein cholesterol levels[7,8], but contribute little to the improvement of tissue insulin sensitivity and acceleration of glucose metabolism. Therefore, novel dual PPARα/γ agonists have received increasing attention given that they might enhance the sensitivity of target tissues to insulin via PPARγ activation and improve lipid metabolic disorders via PPARα activation. In the present paper, we focused on developing a novel compound, C333H (2-(3-furan-2-yl-acryloylamino)-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; Figure 1), which has been shown to be a potent dual PPARα/γ agonist.
Materials and methods
Materials Human normal hepatic LO2 cells were kindly provided by Prof Xiao-ming YANG (Institute of Radiation Medicine, Beijing). Preadipocytes (3T3-L1 cells) were obtained from the Institute of Geriatrics, Beijing Hospital. Mouse myogenic C2C12 cells were purchased from the Cell Center of the Chinese Academy of Medical Sciences. Eight-week-old male homozygous db/db mice were obtained from the Model Animal Research Center of Nanjing University (Grade II, Certificate no SCXK Su 2005-0002).
Oil red O, insulin and fenofibrate were purchased from Sigma (Sigma-aldrich Inc, St Louis, MO). Trizol was the product of Gibco(GIBCO-BRL, Grand Island, NY). The polymerase chain reaction (PCR) primers were synthesized by TaKaRa (TaKaRa Biotechnology Co, Ltd Dalian, China). C333H and rosiglitazone were synthesized in our laboratory. These compounds were dissolved in dimethyl sulfoxide (Me2SO) to prepare the stock solution.
Reporter plasmids and luciferase assays To generate fusion protein expression vectors (pM-PPARα and pM-PPARγ) containing residues 1–147 of the GAL4 DNA-binding domain (DBD) and residues 204–505 of the human PPARγ ligand-binding domain (LBD), or residues 167–467 of the human PPARα LBD, the LBD of hPPARγ and hPPARα from human adipose tissue or liver cDNA libraries were amplified by PCR and subcloned into the pM mammalian expression plasmid. The GAL4-responsive reporter plasmid [pUAS(5x)-tk-luc] containing 5 copies of the GAL4 response element was placed adjacent to the thymidine kinase (tk) minimal promoter and the luciferase reporter gene. pRL-CMV-Rluc (Promega, USA), a reporter vector containing Renilla luciferase (Rluc), was used as an internal control for normalizing transfection efficiency.
HEK-293 cells (human embryonic kidney cells) were seeded at 1.0×104 cells/well in 96-well plates in RPMI-1640 medium containing 10% charcoal-stripped fetal calf serum at 37 ºC in 5% CO2. Transfections were performed followed by 48-h incubation. Specifically, pM-hPPARγ or pM-hPPARα (0.2 µg/well), pUAS(5x)-tk-luc (0.2 µg/well) and pRL-CMV-Rluc were transfected into HEK-293 cells by using the Lipofectamine 2000 system (Invitrogen, USA). After transfection, the cells were cultured for 24 h, and compounds for ligand assay were added into the medium at appropriate concentrations. Following an additional 24-h incubation, the cells were lysed and analyzed by using a dual-luciferase reporter gene assay system (Promega, USA).
Adipocyte differentiation assay The 3T3-L1 cells were cultured in maintenance medium [Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and 10 mg/mL penicillin and streptomycin] at 37 °C in 5% CO2. After two days, cells were treated with the compounds or vehicle (0.05% Me2SO) in the presence of 10 µg/mL insulin every other day. After 7 d, cells were fixed with 10% formaldehyde for 1 h and then stained with oil red O (0.1 mg/mL) for 2 h at room temperature. Medium in each well was then removed, and isopropyl alcohol was added to dissolve the precipitate. The optical density (OD) at a wavelength of 510 nm was determined by enzyme-linked immunosorbent assay (ELISA) spectrometry.
Cell culture Human normal hepatic LO2 cells were cultured in maintenance medium (RPMI -1640 supplemented with 10% FBS) at 37 °C in 5% CO2. Mouse myogenic C2C12 cells were cultured in maintenance medium (DMEM supplemented with 10% FBS) at 37 °C in 5% CO2. When cells were grown to confluency, the media for the C2C12 cells was replaced with differentiation medium (DMEM supplemented with 2% horse serum).
For drug assays, LO2 cells were treated with 10 µmol/L C333H for 24 h, and 3T3-L1 cells were treated with 10 µmol/L C333H in the presence of 10 μg/mL insulin for 72 h. C2C12 cells were differentiated into myotubes for 4 d, and the medium was replaced with phenol red-free differentiation medium supplemented with 10 µmol/L C333H for 24 h.
RT-PCR Total RNA from cells was isolated using Trizol reagent following the manufacturer’s instructions. The RNA content was quantified by using an ultraviolet spectrophotometer at 260 nm. For RT-PCR analysis of hACO, mLPL, maP2 and mGluT4 expression, total RNA was reverse transcribed and subsequently amplified by PCR using the BcaBEST RNA PCR kit (version 1.1; TaKaRa Biotechnology, Dalian, China). The primers for hACO (362 bp product; sense 5'-GGGCATGGCTATTCTCATTGC-3', antisense 5'-CGAA-CAAGGTCAACAGAAGTTAGGTTC-3'); mLPL (403 bp product; sense 5'-CTTTG AGAAAGGGCTCTGCC-3', anti-sense 5'-CCTCTCGATGACGAAGCTGG-3'); maP2 (160 bp product; sense 5'-AAGACAGCTCCTCCTCGAAGGTT-3', antisense 5'-TGACCAAATCCCCATTTACGC-3'); mGluT4 (504 bp product; sense 5'-AACGAGCTGGACGACGGACA-3', antisense 5'-TTGCCCCTC AGTCATTCTCA-3') and the internal control hGAPDH (176 bp product; sense 5'-ACCCA-CTCCTCCACCTTT G-3', antisense 5'-CTCTTGTGCTCTT-GCTGGG-3'); mGAPDH (505 bp product; sense 5'-CCCTGGCC AAGGTCATCCAT-3', antisense 5'- AGGTCCACCACCCTG-TTGCT-3'). The PCR conditions were as follows: 25 (mLPL, mGAPDH, and hGAPDH), 28 (maP2), or 30 (mGluT4 and hACO) cycles of 94 °C for 20 s, 52 °C (mGluT4) or 56 °C (mLPL, maP2, mGAPDH, hGAPDH, and hACO) for 30 s, and 72 °C for 1 min. Following amplification, 5 µL of each PCR product was separated on a 1% agarose gel, stained with ethidium bromide and visualized under ultraviolet light with a MultiImage light cabinet (AlphaImager 2200, USA).
Animal assays Eight-week-old male homozygous db/db mice were treated once daily with 10 mg/kg C333H or 0.5% sodium carboxymethylcellulose (control) by intragastric gavage. C333H was suspended in 0.5% sodium carboxy-methylcellulose. Blood was taken from the retroorbital sinuses at d 0 and d 14 from fasting mice. The various serum parameters were determined by using commercial kits (Rongsheng Biotech, Shanghai, China).
Statistical analysis Data are shown as mean±SD. Differences between individual groups were analyzed by using the t-test for adipocyte differentiation or ANOVA for the animal assay.
Results
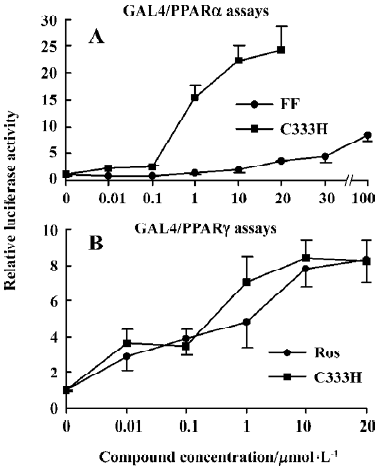
Activation effect of C333H on human PPARα and PPARγ We tested the activation effects of rosiglitazone, fenofibrate, and C333H on human PPARα and PPARγ by using a transient transfection assay. To minimize background noise caused by endogenous PPAR ligands, an established chimera system was used, which contained the yeast GAL4 DBD linked to the LBD of PPARα or PPARγ[9]. Interestingly, rosiglitazone was the only strong activator of PPARγ, and fenofibrate did not have a significant effect on PPARα. C333H was a potent activator of both PPARα and PPARγ. The addition of 10 µmol/L C333H strongly upregulated luciferase activity 22.2-fold for pM-PPARα, whereas 10 µmol/L rosiglitazone (PPARγ agonist) and 100 µmol/L fenofibrate (PPARα agonist) produced only a 3.6-fold (data not shown) and a 8.5-fold increase, respectively. For pM-PPARγ, C333H and rosiglitazone upregulated luciferase activity 8.3- and 8.4-fold, respectively (Figure 2).
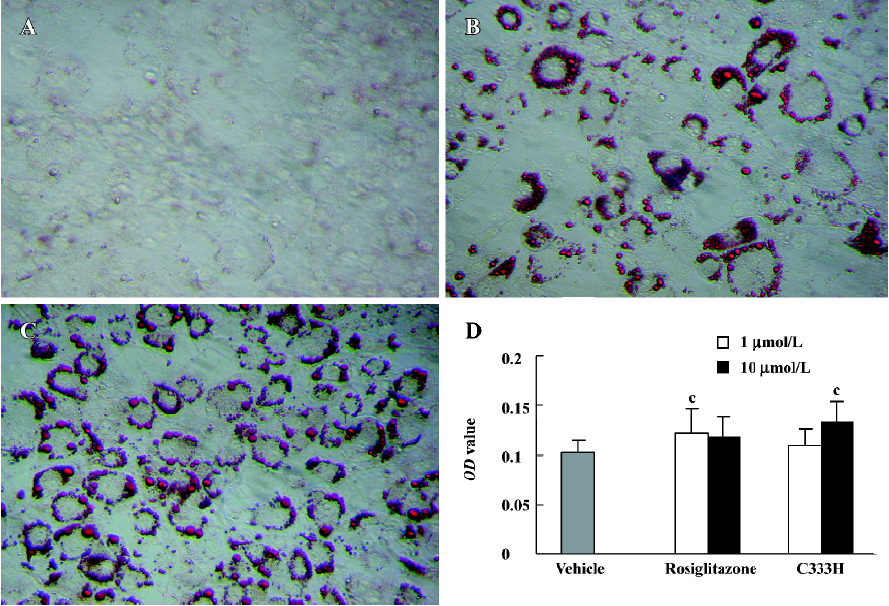
Effect of C333H on adipocyte differentiation It has previously been shown that PPARγ agonists are dominant regulators of adipocyte development[10]. In the present study we found that C333H and rosiglitazone could promote the adipocyte differentiation of 3T3-L1 cells. At the same concentration (10 µmol/L), C333H was a markedly more potent and efficacious inducer of adipogenesis than rosiglitazone (Figure 3).
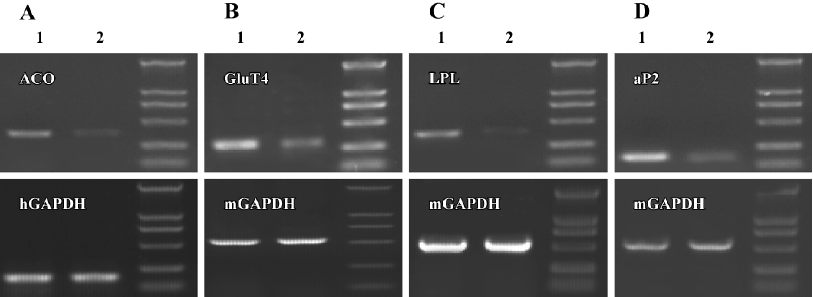
Effect of C333H on gene expression in several cell lines We investigated the regulation of ACO, LPL, aP2, and GluT4 gene expression by C333H in hepatocytes, adipocytes, and skeletal muscle cells. We found that C333H increased the expression levels of ACO mRNA in human LO2 normal hepatocytes, GluT4 in C2C12 skeletal muscle cells, and LPL and aP2 in 3T3-L1 preadipocytes (Figure 4).
Effect of C333H on circulating lipid and glucose levels in db/db mice We investigated the pharmacological effect of C333H in db/db mice. The triglyceride (TG), total cholesterol (T-CHO), free fatty acid (FFA) and glucose serum concentrations were measured at d 0 and d 14 . After 14-d treat-ment with C333H, serum TG, T-CHO, FFA, and glucose were markedly reduced (Table 1).
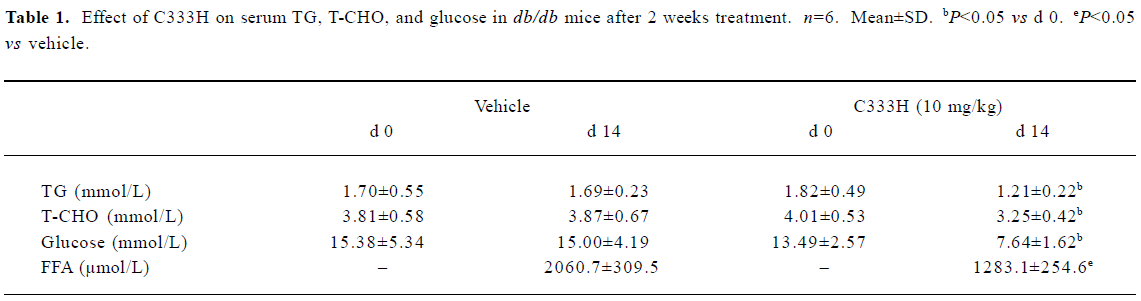
Full table
Discussion
It is known that PPARγ activation improves insulin resistance, and that PPARα activation induces a decrease in circulating lipid levels[11–13]. Rosiglitazone is a potent agonist of PPARγ, whereas its activity with respect to PPARα is weak (data not shown). In vitro reporter gene assays established that C333H was an effective activator of both PPARα and PPARγ. C333H was a more potent agonist of PPARα than fenofibrate, and it was found to have a similar PPARγ activation effect to rosiglitazone. In vivo, we found that C333H significantly reduced the circulating levels of TG, T-CHO, FFA, and glucose in db/db mice, indicating that it might enhance insulin sensitivity and improve lipid metabolic disorders by activating both PPARα and PPARγ.
In an adipocyte differentiation assay, C333H had the highest lipogenic activity of the compounds tested, which indicates that C333H could improve insulin resistance by PPARγ activation. Because PPARγ plays an important role in the regulation of adipocyte differentiation[14], PPARγ agonists can promote preadipocyte differentiation to adipocytes. However, activation of PPARγ can increase the number of small adipocytes and reduce the number of large adipocytes in white adipose tissues. Because small adipocytes are more sensitive to insulin, an increased number of small adipocytes and a decreased number of large adipocytes in white adipose tissues can alleviate insulin resistance[15]. Furthermore, adipocyte differentiation leads to the expression of adipocyte-specific genes, such as aP2[16], LPL[17], and GluT4[18], which indicates that PPARγ agonists have good antihyperglycemic and antihyperlipidemic activity.
Liver, adipose tissue, and skeletal muscle are major sites of glucose and lipid metabolism. PPARα is predominantly expressed in the liver, whereas PPARγ is most abundant in adipose tissue, and is also expressed in small amounts in skeletal muscle[19,20]. In the present study, the ability of C333H to regulate ACO, LPL, aP2, and GluT4 gene expression was investigated in various cell lines. Hepatic PPARα-dependent ACO mainly regulates fatty acid β-oxidation. LPL and aP2 are regulated by PPARγ, and mainly function in triglyceride and fatty acid metabolism[21]. Moreover, the insulin-dependent glucose transporter GluT4 functions to regulate glucose uptake into adipose tissue and skeletal muscle in response to elevated levels of insulin in the circulation. Our results showed that C333H upregulated the expression levels of these genes, indicating that it could lower blood glucose and lipid concentrations in type 2 diabetic patients.
In summary, we demonstrated that C333H was a dual activator of PPARα and PPARγ. It not only controlled glucose and lipid metabolism, but also promoted preadipocyte differentiation and improved insulin resistance. These results suggest that further studies should be carried out to develop C333H as a novel therapy for metabolic disease such as obesity, hyperlipidemia and type 2 diabetes.
References
- Grundy SM, Barnett JP. Metabolic and health complications of obesity. Dis Mon 1990;36:641-713.
- Ginsberg H, Plutzky J, Sobel E. Review of metabolic and cardiovascular effects of oral antidiabetic agents: beyond glucose level lowering. J Cardivasc Risk 1999;6:337-46.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835-9.
- Schoonjans K, Martin G, Staels B, Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr Opin Lipidol 1997;8:159-66.
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem 2000;43:527-50.
- Bole PJ, King AB, Olansky L, Marchetti A, Lau H, Magar R, et al. Effects of pioglitazone and rosiglitazone on blood lipid level and glycemic control in patients with type 2 diabetes mellitus: a retrospective review of randomly selected medical records. Clin Ther 2002;24:378-96.
- Chaput E, Saladin R, Sliverstre M, Edgar AD. Fenofibrate and rosiglitazone lower serum triglycerides with opposing effects on body weight. Biochem Biophys Res Commun 2000;271:445-50.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998;98:2088-93.
- Sadoeski I, Ptashne M. A vector for expressing GAL4 (1-147) fusion in mammalian cells. Nucl Acids Res 1989;17:7539.
- Tontonoz P, Graves RA, Budavari AI, Erdjument-Bromage H, Lui M, Hu E, et al. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPARγ and RXRα. Nucl Acids Res 1994;22:5628-34.
- Camp HS, Li O, Wise SC. Differential activation of peroxisome proliferator-activated receptor-gamma by troglitazone and rosiglitazone. Diabetes 2000;49:539-47.
- Vazquez M, Silvestre JS, Prous JR. Experimental approaches to study PPARγ agonists as antidiabetic drugs. Methods Find Exp Clin Pharmacol 2002;24:515-23.
- Fruchart JC. Peroxisome proliferator-activated receptor-α activation and high-density lipoprotein metabolism. Am J Cardiol 2001;88:24N-9N.
- Lowell BB. PPARγ: an essential regulator of adipogenesis and modulator of fat cell function. Cell 1999;99:230-53.
- Okuno A, Tamemoto H, Tobe K. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese zucker rats. J Clin Invest 1998;101:1354-61.
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994;79:1147-56.
- Schoonjans K, Peinado-Onsurbe J, Lefebvre AM. PPARα and PPARγ activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 1996;15:5336-48.
- Wu Z, Xie Y, Morrison RF, Bucher NLR, Farmer SR. PPARγ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J Clin Invest 1998;101:22-32.
- Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A. Peroxisome proliferator-activated receptor gene expression in human tissues. J Clin Invest 1997;99:2416-22.
- Kruszynska YT, Mukherjee R, Jow L, Dana S, Paterniti JR, Olefsky JM. Skeletal muscle peroxisome proliferation-activated receptor-γ expression in obesity and non-insulin-dependent diabetes mellitus. J Clin Invest 1998;101:543-8.
- Escher P, Wahli W. Peroxisome proliferator-activated receptor: insight into multiple cellular function. Mutat Res 2000;448:121-38.