
Pranlukast reduces neutrophil but not macrophage/microglial accumulation in brain after focal cerebral ischemia in mice1
Introduction
After cerebral ischemia, the highly complex pathophysiological process that follows can be separated into 3 successive phases: metabolic stress and excitotoxicity (acute, within hours), inflammation and apoptosis (subacute, hours to days), and repair and regeneration (chronic, days to months)[1,2]. Post-ischemic inflammation in the subacute phase is an important event in which a large number of cells and molecules/mediators are involved. Among the inflammatory cells, the accumulation of neutrophils and macrophage/microglia in the brain is a determinant in pathogenesis[3–5]. Cysteinyl leukotrienes (CysLT, including LTC4, LTD4 and LTE4), the 5-lipoxygenase metabolites of arachidonic acid, represent one type of pro-inflammatory mediator involved in cerebral ischemia[6–9]. CysLT can increase blood–brain barrier (BBB) permeability and induce brain edema after cerebral ischemia and neutrophil perfusion[6,9]. However, their actions on inflammatory cells in the ischemic brain are still unknown, although neutrophil-endothelial cell cooperation has been suggested to induce brain edema via production of CysLT in guinea pig brains perfused with human neutrophils[9]. In peripheral tissues, CysLT can increase eosinophil adhesion[10] and transendothelial migration[11], which are involved in airway eosinophilic inflammation in asthma[12].
The pro-inflammatory actions of CysLT are mediated by activating cysteinyl leukotriene receptors (CysLT1 and CysLT2)[13]. CysLT1 receptor mRNA has been detected by Northern blotting in the brain[14], and its protein is primarily expressed in microvascular endothelium in human brain tissues[15]. We previously found that the CysLT1 receptor antagonists pranlukast (ONO-1078) and montelukast protected against acute and chronic ischemic brain injury in rats and mice[16–21]. This effect may partly result from inhibiting BBB permeability, brain edema, and glial scar formation[17,20,21]. However, whether pranlukast inhibits inflammatory cells in ischemic brain tissue is not yet clear.
Therefore, to further determine whether the protective effect of pranlukast is associated with anti-inflammatory activity, in the present study we observed its effect on cerebral ischemia-induced neutrophil and macrophage/microglial accumulation as well as BBB disruption and neuronal injury in mice with focal cerebral ischemia.
Materials and methods
Materials Pranlukast (ONO-1078) was kindly provided by Dr Masami TSUBOSHIMA (Ono Pharmaceutical Co, Osaka, Japan). Chloral hydrate, 2,3,5-triphenyltetrazolium chloride (TTC) and biotinylated anti-mouse IgG antibody were purchased from Sigma (St Louis, USA). Fluoro-Jade B was purchased from Chemicon International (Temecula, CA, USA). Biotinylated anti-rabbit IgG, biotinylated anti-rat IgG, horseradish peroxidase streptavidin and 3,3'-diaminobenzi-dine were purchased from Zhongshan Biotechnology (Beijing, China). Polyclonal rabbit anti-myeloperoxidase (MPO) and monoclonal rat anti-CD11b antibodies were purchased from Serotec (Oxford, UK).
Physiological parameter monitoring Male Kunming mice weighing 25–30 g (Shanghai Experimental Animal Center, China; Certificate N
Mice were anesthetized by intraperitoneal injection of chloral hydrate (400 mg/kg). A polyethylene tube was inserted into the right femoral artery for continuously monitoring blood pressure, by using a computer-assisted system (MedLab-U/4cs; Nanjing MedEase, Nanjing, China), and for measuring PaO2, PaCO2, and arterial blood pH (ABL 330 blood gas analyzer; Leidu, Denmark). Blood glucose was monitored by using a one touch basic blood glucose monitoring system (Lifescan, USA). Rectal (core) temperature was measured and maintained at 37.0±0.5 ºC with a heating pad and a heating lamp during the surgery.
Focal cerebral ischemia Focal cerebral ischemia was induced by permanent middle cerebral artery occlusion (MCAO) as previously described[20]. Briefly, a 6–0 nylon monofilament suture, blunted at the tip and coated with 1% poly-L-lysine, was inserted into the right internal carotid artery, and advanced approximately 10 mm distal to the carotid bifurcation to occlude the origin of the middle cerebral artery. In sham-operated animals, the same procedure was carried out, except that an intraluminal filament was inserted. Pranlukast (0.01 and 0.1 mg/kg) was ip injected 30 min before and 30 min after MCAO.
Neurological deficits and histopathological assessment Neurological deficit scores were evaluated 72 h after MCAO as described by Bederson et al[22]: 0, no deficit; 1, failure to extend left forepaw fully; 2, circling to the left; 3, failing to the left; 4, no spontaneous walking with a depressed level of consciousness.
Mice were re-anesthetized 72 h after MCAO, and brains were quickly removed and dissected into 1.5 mm-thick coronal slices. The slices were stained with 0.5% TTC at 37 oC for 30 min, and then fixed in a 10% buffered formalin solution. The stained slices with the caudal face upward were photographed by a charge coupling device CCD camera (CP 230; Panasonic, Japan) and images were recorded on a computer. Adjusted infarct area and both hemisphere areas of each slice were determined by using an image analysis program (AnalyPower 1.0; Zhejiang University, Hangzhou, China) as reported elsewhere[20]. Infarct volume was calculated as lesion area×thickness (1.5 mm). The summation of the infarct volumes of all brain slices was the total infarct volume.
In another series, mice were deeply anesthetized 72 h after MCAO, and perfused transcardially with saline followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4). The brains were removed, and fixed overnight in the same fixative as described earlier, and then immersed in 30% sucrose solution in phosphate buffer. The brains were frozen and 20 µm- or 10 µm-thick coronal sections were cut by cryomicrotomy (CM1900; Leica, Germany). The 10 µm-thick sections were used for immunohistochemical analysis, and the 20 µm-thick sections were stained with 0.0004% Fluoro-Jade B to detect degenerated neurons by fluorescent microscopy[23].
Immunohistochemical analyses To determine neutrophil and macrophage/microglial accumulation, the brain sections were immunostained with anti-MPO (marker of neutrophils)[24] or anti-CD11b (marker of macrophage/microglia) antibodies[25]. The 10 μm-thick sections were sequentially incubated with 5% goat serum for 2 h, rabbit polyclonal anti-MPO (1:200) or rat monoclonal anti-CD11b (1:200) antibodies overnight, biotinylated anti-rabbit and anti-rat IgG (1:200) for 2 h, and streptavidin avidin-biotin-horseradish peroxidase complex (1:200) for 2 h. Finally, the sections were exposed for 5–20 min to 0.01% 3,3'-diaminobenzi-dine.
Endogenous IgG immunostaining was performed to detect BBB disruption[26]. Brain sections were reacted respectively and successively with biotinylated anti-mouse IgG antibody (1:500), horseradish peroxidase streptavidin (1:200) and 3,3'-diaminobenzidine. The gray scales in the immuno-stained sections were detected with an image analyzer (Imagetool 2.0; University of Texas Health Science Center, San Antonio, TX, USA). IgG exudation was evaluated as the percentage increase of the gray scale in the ischemic hemisphere: (Gi–G0)/G0×l00%, where Gi is the gray scale of the ischemic hemisphere and G0 is the gray scale of the contralateral non-ischemic hemisphere.
Statistical analysis Values are presented as mean±SD. One-way ANOVA (Student-Newman-Keuls) or the nonparametric Mann-Whitney U-test was used for statistical analysis using the SPSS software package (version 10.0 for Windows; SPSS, USA). P<0.05 was considered statistically significant.
Results
There were no significant differences in mean arterial blood pressure, or arterial blood PaO2, PaCO2, or blood glucose between 30 min before and 30 min after MCAO among the groups treated with saline and pranlukast (0.01 and 0.1 mg/kg) as well as those animals that received a sham operation (Table 1).
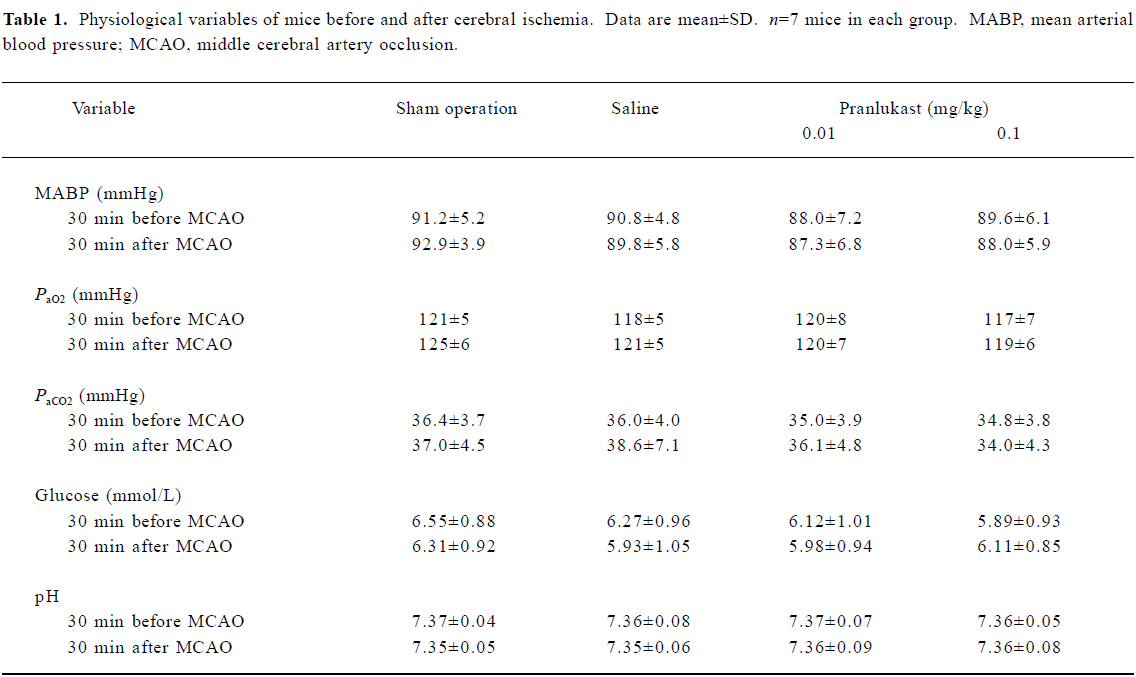
Full table
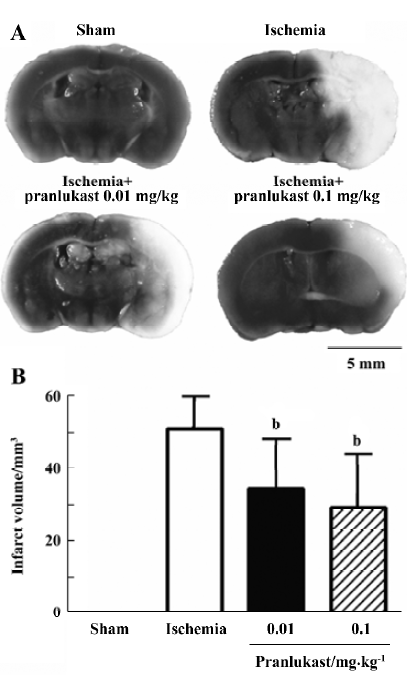
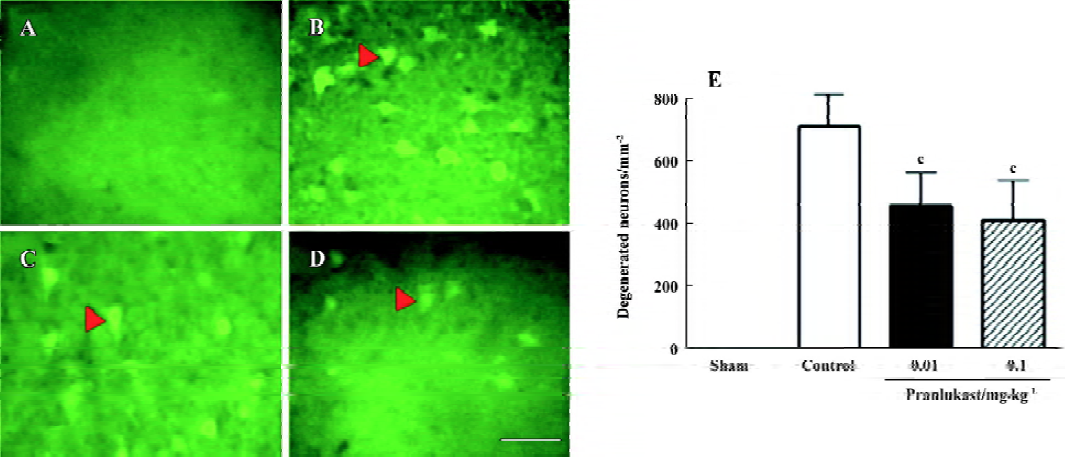
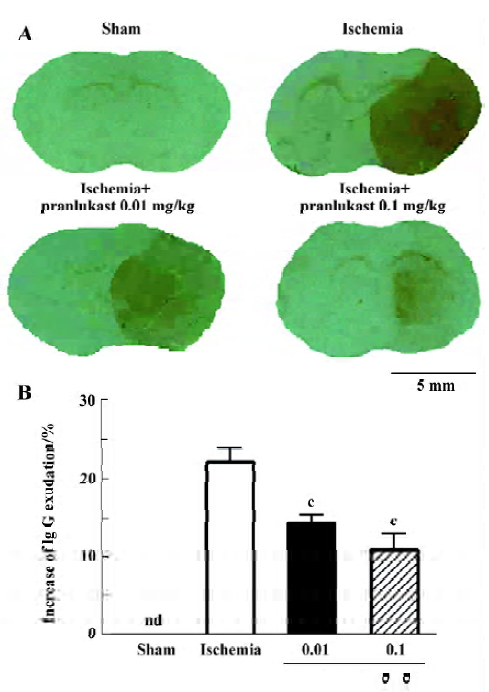
Focal cerebral ischemia induced neurological deficits (2.20±0.71, n=16) 72 h after MCAO. Pranlukast reduced the neurological deficit score by 20% at a dose of 0.01 mg/kg (1.76±0.75; n=16, P>0.05 vs ischemic control; Mann-Whitney U-test) and by 37% at 0.1 mg/kg (1.38±0.57; n=16, P<0.05 vs ischemic control) 72 h after MCAO. Pranlukast at doses of 0.01 and 0.1 mg/kg significantly decreased the infarct volume by 32.2% and 42.9%, respectively, 72 h after MCAO (P<0.05 vs ischemic control; Figure 1). The number of Fluoro-Jade B-positive cells (degenerated neurons) in the temporoparietal cortex III and IV layers was increased 72 h after MCAO, and significantly reduced by pranlukast (0.01 and 0.1 mg/kg; Figure 2). In addition, endogenous IgG immunoreactivity was increased by 22.2%±4.2% in the ischemic hemisphere as compared with the contralateral non-ischemic hemisphere, and pranlukast (0.01 and 0.1 mg/kg) markedly reduced IgG exudation (Figure 3).
The numbers of MPO-positive neutrophils and CD11b-positive macrophages/microglia were increased in the ischemic boundary region 72 h after MCAO (Figures 4 and 5). Pranlukast (0.01 and 0.1 mg/kg) significantly inhibited neutrophil accumulation (Figure 4C, 4D), but did not inhibit macrophage/microglial accumulation (Figure 5C, 5D).
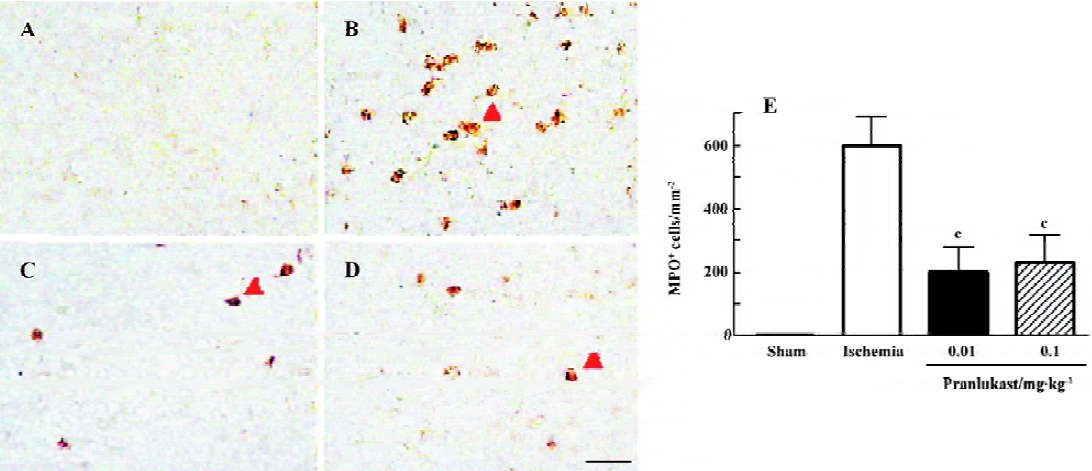
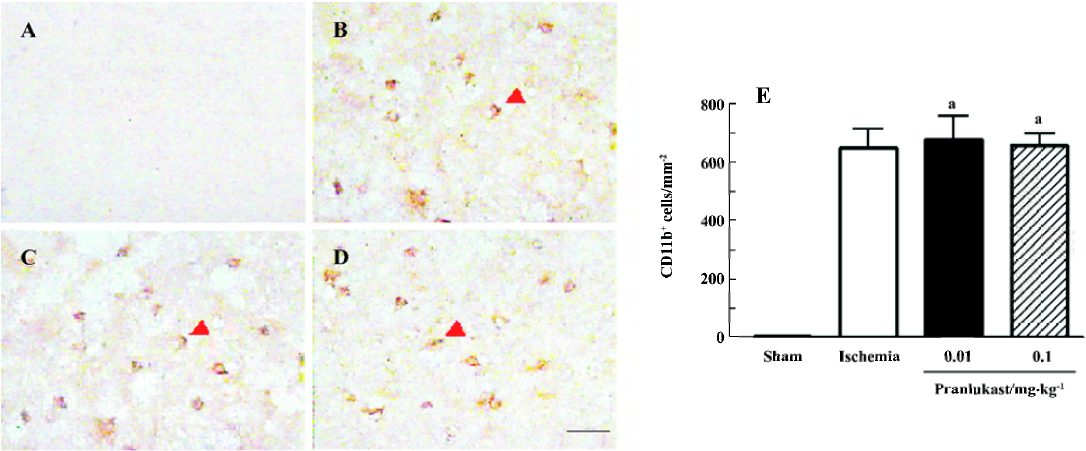
Discussion
In the present study, the most important finding is that pranlukast inhibited neutrophil but not macrophage/microglial accumulation in ischemic brain tissue 72 h after MCAO in mice, in addition to ameliorating neurological deficits and neuron degeneration and reducing infarct volume and IgG exudation. These results not only further confirm the neuroprotective effect of pranlukast on focal cerebral ischemia as reported elsewhere[16–21], but also demonstrate its anti-inflammatory properties.
The effect of pranlukast on post-ischemic neutrophil accumulation might result from the inhibition of BBB disruption. The present results indicate that pranlukast inhibits endogenous IgG exudation, an indicator of BBB disruption, as found in previous studies through endogenous plasma albumin or Evans blue staining[16,17,20]. Because neutrophils are hematogenous inflammatory cells, their accumulation in ischemic brain tissues may reflect BBB disruption because the disrupted BBB has been reported to promote blood leukocyte recruitment[2,5].
The lack of effect of pranlukast on post-ischemic macrophage/microglial accumulation might indicate that CysLT1 receptor is not involved in this response in the subacute phase (72 h after MCAO). Resident microglia and hematogenous macrophages play similar roles in the pathogenetic cascade following cerebral ischemia, and both of them are CD11b-positive, but distinction between these cells has not been possible due to a lack of discriminating cellular markers[25,27]. However, in mice transplanted with green fluorescent protein (GFP)-transgenic bone marrow, hematogenous GFP+ macrophages were rarely observed on d 2, reached peak numbers on d 7, and decreased thereafter; in contrast, resident GFP– microglia rapidly became activated on d 1 after MCAO[27]. Therefore, most CD11b-positive cells 72 h after MCAO in the present study might be activated resident microglia. Although the activation of microglia in the subacute phase might not be regulated by CysLT1 receptor, further observations are needed, because this receptor may be expressed in later phases, and many other factors may influence the effect of pranlukast.
In summary, pranlukast inhibits post-ischemic inflammation in the brain, mainly acting on the recruitment of hematogenous inflammatory cells (such as neutrophils) via inhibiting BBB disruption. This finding is consistent with the localization of CysLT1 receptor in microvascular endothelium in the human brain[15]. The anti-inflammatory effects of pranlukast have been reported in peripheral tissues, for example allergen-induced interleukin-5 production in human lung tissue[28], transendothelial migration of eosinophils across human umbilical vein endothelial cells in response to LTD4[11], and eosinophil adhesion[10]. In the present paper, we demonstrated the anti-inflammatory effect of pranlukast in the focal ischemic brain, but this effect might be limited to BBB disruption-associated neutrophil recruitment. Thus, the present study indicates that the anti-inflammatory effect of pranlukast may be partly involved in its neuroprotective effect against cerebral ischemic injury.
Acknowledgement
We thank Dr Masami TSUBOSHIMA, Ono Pharmaceutical Co, Osaka, Japan, for supplying the pranlukast.
References
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003;26:248-54.
- Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke 2004;35:2220-5.
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003;62:127-36.
- Simundic AM, Basic V, Topic E, Demarin V, Vrkic N, Kunovic B, et al. Soluble adhesion molecules in acute ischemic stroke. Clin Invest Med 2004;27:86-92.
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 2005;289:H558-68.
- Baba T, Black KL, Ikezaki K, Chen KN, Becker DP. Intracarotid infusion of leukotriene C4 selectively increases blood-brain barrier permeability after focal ischemia in rats. J Cereb Blood Flow Metab 1991;11:638-43.
- Rao AM, Hatcher JF, Kindy MS, Dempsey RJ. Arachidonic acid and leukotriene C4: role in transient cerebral ischemia of gerbils. Neurochem Res 1999;24:1225-32.
- Ciceri P, Rabuffetti M, Monopoli A, Nicosia S. Production of leukotrienes in a model of focal cerebral ischaemia in the rat. Br J Pharmacol 2001;133:1323-9.
- Di Gennaro A, Carnini C, Buccellati C, Ballerio R, Zarini S, Fumagalli F, et al. Cysteinyl-leukotrienes receptor activation in brain inflammatory reactions and cerebral edema formation: a role for transcellular biosynthesis of cysteinyl-leukotrienes. FASEB J 2004;18:842-4.
- Nagata M, Saito K, Tsuchiya K, Sakamoto Y. Leukotriene D4 upregulates eosinophil adhesion via the cysteinyl leukotriene 1 receptor. J Allergy Clin Immunol 2002;109:676-80.
- Nagata M, Saito K, Kikuchi I, Hagiwara K, Kanazawa M. Effect of the cysteinyl leukotriene antagonist pranlukast on trans-endothelial migration of eosinophils. Int Arch Allergy Immunol 2005;137:S2-6.
- Nagata M, Saito K. The roles of cysteinyl leukotrienes in eosinophilic inflammation of asthmatic airways. Int Arch Allergy Immunol 2003;131:S7-10.
- Brink C, Dahlen SE, Drazen J, Evans JF, Hay DW, Nicosia S, et al. International Union of Pharmacology XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol Rev 2003;55:195-227.
- Sarau HM, Ames RS, Chambers J, Ellis C, Elshourbagy N, Foley JJ, et al. Identification, molecular cloning, expression, and characterization of a cysteinyl leukotriene receptor. Mol Pharmacol 1999;56:657-63.
- Zhang WP, Hu H, Zhang L, Ding W, Yao HT, Chen KD, et al. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci Lett 2004;363:247-51.
- Zeng LH, Zhang WP, Wang RD, Wang PL, Wei EQ. Protective effect of ONO-1078, a leukotriene antagonist, on focal cerebral ischemia in mice. Yao Xue Xue Bao 2001;36:148-50. Chinese..
- Zhang WP, Wei EQ, Mei RH, Zhu CY, Zhao MH. Neuroprotec-tive effect of ONO-1078, a leukotriene receptor antagonist, on focal cerebral ischemia in rats. Acta Pharmacol Sin 2002;23:871-7.
- Zhang LH, Wei EQ. Neuroprotective effect of ONO-1078, a leukotriene receptor antagonist, on transient global cerebral ischemia in rats. Acta Pharmacol Sin 2003;24:1241-7.
- Zhang SH, Wei EQ, Zhu CY, Chen Z, Zhang SF. Protective effect of ONO-1078, a leukotriene receptor antagonist, on focal cerebral ischemia induced by endothelin-1 in rats. Yao Xue Xue Bao 2004;39:1-4. Chinese..
- Yu GL, Wei EQ, Zhang SH, Xu HM, Chu LS, Zhang WP, et al. Montelukast, a cysteinyl leukotriene receptor-1 antagonist, dose- and time-dependently protects against focal cerebral ischemia in mice. Pharmacology 2005;73:31-40.
- Yu GL, Wei EQ, Wang ML, Zhang WP, Zhang SH, Weng JQ, et al. Pranlukast, a cysteinyl leukotriene receptor-1 antagonist, protects against chronic ischemic brain injury and inhibits the glial scar formation in mice. Brain Res 2005;1053:116-25.
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 1986;17:472-6.
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000;874:123-30.
- Yang GY, Liu XH, Kadoya C, Zhao YJ, Mao Y, Davidson BL, et al. Attenuation of ischemic inflammatory response in mouse brain using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist. J Cereb Blood Flow Metab 1998;18:840-7.
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA 1991;88:7438-42.
- Jensen MB, Finsen B, Zimmer J. Morphological and immuno-phenotypic microglial changes in the denervated fascia dentata of adult rats: correlation with blood-brain barrier damage and astroglial reactions. Exp Neurol 1997;143:103-16.
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25-33.
- Fukushima C, Matsuse H, Hishikawa Y, Kondo Y, Machida I, Saeki S, et al. Pranlukast, a leukotriene receptor antagonist, inhibits interleukin-5 production via a mechanism distinct from leukotriene receptor antagonism. Int Arch Allergy Immunol 2005;136:165-72.