
Targeting leukemic fusion proteins with small interfering RNAs: recent advances and therapeutic potentials1
Introduction
The discovery of RNA interference in 1998 by Fire and coworkers provided an unanticipated new approach for studying gene functions in many different cell types and species from various kingdoms[1,2]. RNA interference (RNAi) is induced by exogenously introduced double-stranded RNAs (dsRNAs), RNA viruses, transposons and endo-genous short dsRNAs, and may be an ancient defense system against viral infections and other genetic invaders[3]. The term RNA interference was first introduced in 1998, after the discovery by Andrew Fire and collaborators that injection of long dsRNA into Caenorhabditis elegans led to efficient and specific gene silencing[1]. However, in contrast to the situation in invertebrates, introduction of dsRNAs longer than 30 base pairs into mammalian somatic cells activates interferon responses, finally leading to a general inhibition of gene expression. Nevertheless, the identification of small interfering RNAs (siRNAs), double-stranded RNAs of 21 to 28 nucleotides in length, as intermediates of the RNAi process paved the way for applying RNAi in mammalian cells[4]. Because of their short lengths, siRNAs only rarely induce interferon responses, but cause an efficient and sequence-specific inhibition of gene expression. An increasing number of vector systems for the ectopic expression of small hairpin RNAs (shRNAs), the intracellularly expressed cousins of siRNAs, complement improved chemical synthesis of siRNAs for transient applications. All these advancements have allowed RNA interference to become a standard tool in molecular and cellular biology within 3 years. Moreover, both siRNAs and shRNAs have been successfully applied in vivo in animal models. Because of its high reproducibility, specificity and efficacy, in combination with rapid advances in the delivery of RNAi-inducing molecules, RNAi holds great promise for the development of new therapeutic strategies to combat diseases such as cancer[5,6].
Leukemia is a malignant disease of the hematopoietic system. Leukemic cells are characterized by impaired differentiation and increased proliferation potential, leading to an expansion of the leukemic clone and the replacement of normal hematopoiesis. In contrast to most types of solid tumors, more than 50% of all leukemia cases are associated with distinct chromosomal changes, such as translocations or inversions. Moreover, leukemias tend to exhibit a higher genetic stability than solid tumors. For these 2 reasons, molecularly defined approaches targeting tumor-specific genes such as fusion genes may be more promising for leukemias than for solid tumors.
The leukemic fusion genes generated by chromosomal translocations are hallmarks of human leukemias. Prominent examples are the BCR-ABL fusion gene, which is associated with chronic myeloid leukemia and acute lymphoblastic leukemia (ALL), and AML1/MTG8, which is involved in acute myeloid leukemia (AML). Kinase fusion genes such as BCR-ABL or TEL-PDGFβR directly disturb signal transduction pathways and provide proliferative and survival advantages. Chimeric transcription factors such as AML1/MTG8, PML-RARα or MLL-AF4 affect histone and DNA modifications and impair differentiation and induce cell death. If such fusion genes still played central roles in the maintenance of leukemia, they could be very promising siRNA targets for molecularly defined treatments, thereby complementing established treatment protocols or new, small molecular drug-based strategies such as inhibition of histone deacetylation or DNA methylation[7–9].
Here, we summarize a selection of such approaches to knockdown leukemic fusion proteins, and discuss the possible applications of RNAi in the development of new therapies for leukemic diseases. A overview of several fusion transcript-specific siRNAs is given in Table 1.

Full table
Short overview of the siRNA mechanism
Naturally occurring siRNAs are generated from long double-stranded RNAs by the RNase III-type enzyme Dicer. The cleavage creates 5'-phosphate and 3'-hydroxyl termini and yields 21–28 long double stranded RNAs with 2 nucleotide-long 3'-overhangs[10]. The siRNAs associate then with a multiprotein complex to form the RNA-induced silencing complex (RISC; Figure 1). RISC is activated by unwinding the siRNA duplex and discarding one of the strands. Recent data suggest that a Dicer-containing protein complex may also facilitate these 2 steps[11]. The remaining strand guides RISC to complementary RNA sequences. The RISC component AGO2 cleaves the target sequence 11 nucleotides away from the 5'-end of the siRNA, leading to the rapid degradation and, thus, inactivation of the target transcript[12,13]. Alternatively, particularly in the case of imperfect homology between target sequence and guide strand, RISC may not cleave the target transcript, but may instead inhibit its translation.
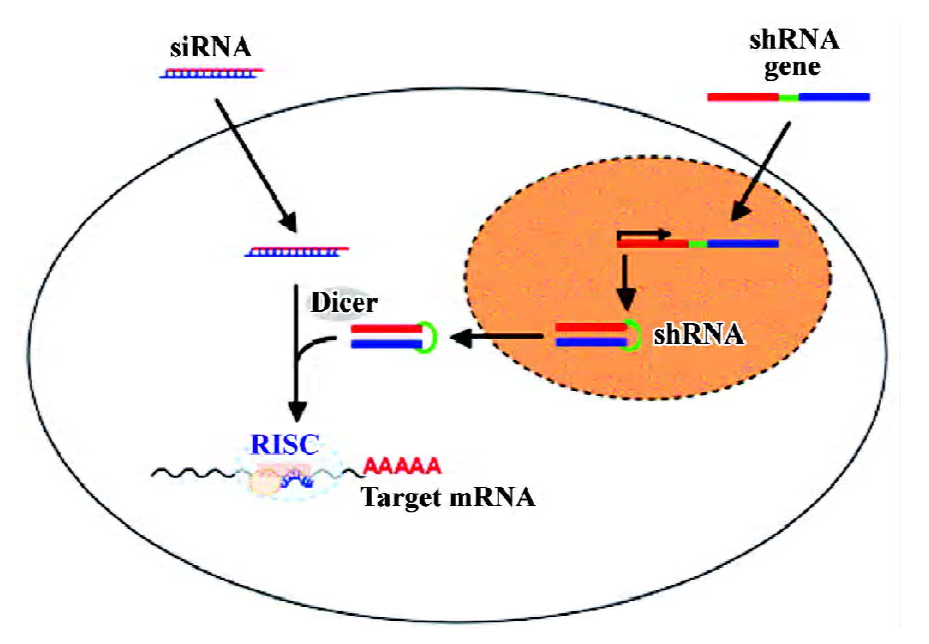
Translational interference without degradation of the transcript is also a feature of endogenously expressed micro RNAs (miRNAs). The eminent roles of miRNAs in the regulation of gene expression and the consequences for cellular processes such as differentiation or proliferation have only just recently come to the fore. In contrast to siRNAs, animal miRNAs have an imperfect homology to their binding sequence, which seems to be responsible for the inability of miRNAs to trigger RNA cleavage[14]. Nevertheless, despite differences in the mechanism (RNA degradation versus inhibition of translation), siRNAs and miRNAs share protein components to form the corresponding nucleoprotein complexes[15].
Both siRNA-mediated RNA degradation and miRNA-mediated inhibition of translation take place in the cyto-plasm[16]. However, RISC does not seem to freely diffuse through the cytoplasm, but has been shown to be part of the cytoplasmic P-bodies[17,18]. Thus, post-transcriptional gene silencing takes place in defined structures, like many other essential cellular processes.
siRNA design and delivery ex vivo
The efficiency and specificity of an siRNA is dependent on both siRNA and target site properties. One important point to consider is the choice of the guide strand by RISC. Strand selection is controlled by the thermodynamic stability of the siRNA termini. The strand with the lower 5'-terminal thermodynamic stability is more likely to stay with RISC. Furthermore, an adenosine residue at position 11 of the antisense strand, the complementary position of the cleavage site, can be advantageous for cleavage efficiency, as RISC prefers to cleave RNA to the 3' side of a uridine. However, these rules are not absolute, and siRNAs, which do not adhere to these rules, do not necessarily have inferior activity. Targeting the fusion site of a leukemic fusion transcript may, depending on the sequences flanking this site, not allow these rules to be strictly obeyed. Instead, a compromise sequence must be found, or, if this is not possible, “non-consensus” siRNAs can be tried. For instance, we identified active siRNAs by scanning a leukemic fusion site with several siRNAs. With this approach, we obtained active and specific siRNAs, which did not follow any of the rules mentioned here (Figure 2)[19].

Transient transfections of siRNA duplexes into mammalian cells can be performed with various commercially available cationic lipid formulations, or with electroporation[20,21]. The advantage of such a transient approach is the lack of cell adaptation to reduced target protein levels. Furthermore, genes essential for cell proliferation can be targeted by transient siRNA approaches. However, there is no single transfection method that can be successfully applied to all cell types under all experimental conditions. It is therefore important to optimize transfection conditions so that maximum gene silencing is achieved. The following transfection parameters have been shown to affect transfection and gene silencing efficacy: cell culture conditions, including cell density and medium composition; the type and amount of transfection agent; the quality and quantity of siRNA; and the length of time that the cells are exposed to the siRNA. Differences have been reported in the ability to transfect and silence gene expression between adherent and nonadherent cells. Post mitotic cells such as neurons and muscle cells tend to be more difficult to transfect using liposomes compared with mitotic cells such as stem cells, fibroblasts and tumor cells.
A major disadvantage of transfections using preformed siRNA lies directly in their transient nature: a long-lived protein may not be affected by such an approach. To overcome this limitation, several siRNA expression systems based on plasmid and viral vectors have been developed to achieve a sustained “knockdown” of the gene of interest[22]. In most cases, the expression cassettes contain an RNA polymerase III-dependent promoter such as the U6 or the H1 promoters. The siRNAs are expressed as hairpin structures known as short hairpin RNAs (shRNAs), which are processed by Dicer to the mature siRNAs[23].
Because the application of plasmid vectors is frequently hampered by inefficient stable transfection rates and by silencing of the shRNA genes, viral vector systems for the expression of shRNAs have been developed. In particular, lentiviral and oncoretroviral systems have been demonstrated to be effective in most cell lines and primary cell types. Because lentiviruses are able to infect non-cycling and post-mitotic cells, they are now widely used to establish a stable RNAi in, for example, neuronal cells, stem cells and transgenic mouse models[24].
Targeting kinases with siRNAs
BCR-ABL The vast majority of all chronic myeloid leukemia (CML) cases and a significant fraction of ALL cases are associated with a chromosomal translocation t(9;22)(q34;q11), also known as the Philadelphia chromosome. This translocation involves the reciprocal transfer of the 3' Abelson proto-oncogene (ABL1) sequence of chromosome 9 to variable locations in the 3'-breakpoint cluster region (BCR) of chromosome 22. The resulting fusion gene BCR-ABL is transcribed into 2 differently spliced chimeric mRNA encoding p210 or p190 BCR-ABL proteins. Both isoforms exhibit constitutively active tyrosine kinase activity. The development of the BCR-ABL rearrangement in hemopoietic stem cells (HSC) is the determining event for the development of CML, leading to a cell clone with a competitive growth advantage over the normal stem cells[25].
A major breakthrough in the treatment of BCR-ABL-associated leukemia was the therapeutic application of the tyrosine kinase inhibitor imatinib mesylate (STI571, Gleevec). Administration of this drug results in marked clinical and cytogenetic remissions. However, this compound is not perfectly specific for BCR-ABL, but also inhibits the tyrosine kinase activities of ABL1, KIT and PDGFR. Moreover, and more importantly, clinical resistance due to point mutations in the ABL kinase domain of BCR-ABL leading to an increasing population of drug-resistant patients is frequently observed[26,27]. These limitations necessitate the development of alternative strategies to complement existing treatment protocols.
Different approaches have been used to deactivate the chimeric protein, such as single-stranded antisense oligonucleotides, which, however, yielded conflicting results with respect to specificity and functionality[28]. To overcome this, several groups used siRNAs to target BCR-ABL mRNA as an alternative approach[29–31]. Consistently, such siRNAs decreased BCR-ABL expression without affecting BCR and ABL1 expression. BCR-ABL depletion was associated with a decreased leukemic proliferation and an increased extent of apoptosis. Despite the fact that cell lines were used in these studies, they provide a basis for a possible siRNA application not only for functional analysis but also as a therapeutic tool. The potential therapeutic value of a lentiviral gene transfer strategy was evaluated as an alternative to the exogenous delivery of chemically synthesized siRNAs[32,33]. The resultant stable expression of BCR-ABL shRNAs yielded a sustained reduction of BCR-ABL, and, consequently, inhibited proliferation, decreased cell survival and compromised colony formation of CD34+ CML cells. Withey et al targeted BCR-ABL in primary chronic phase CML[34]. The siRNA treatment inhibited the expansion of granulocyte-macrophage progenitors expressing the b3a2 variant of BCR-ABL without having any obvious effects on cells expressing the b2a2 variant. This study underlines the role of BCR-ABL in driving aberrant myeloid progenitor amplification.
Baba and colleagues targeted an siRNA against a downstream sequence in the ABL portion instead of the fusion site[35,36]. Thus, this siRNA has the potential to interfere with both BCR-ABL and ABL1 expression. They demonstrated that this siRNA markedly decreased target mRNA levels, consequently leading to the inhibition of protein tyrosine kinase activity and suppression of cell proliferation. Microarray analysis revealed cross talk between siRNA-mediated suppression of the BCR-ABL oncogene and expression of several apoptotic/antiapoptotic and cell proliferation factors. Bartova and colleagues studied the dependence of nuclear topography and expression of the BCR-ABL fusion gene in the leukemic cell line K562 on the expression of nonfused ABL1[37]. They showed that not only BCR-ABL suppression but also inhibition of ABL expression induced downregulation of BCR-ABL and ABL1 proteins, which appeared to be sufficient for the stimulation of apoptosis.
Of the particular interest is the work of Wohlbold and collaborators, who demonstrated that combined treatment with BCR-ABL siRNAs together with imatinib mesylate could overcome a partial imatinib mesylate resistance[38]. In this case, decreasing the quantity of BCR-ABL protein using siRNA could antagonize the 2 major mechanisms of imatinib mesylate resistance, namely overexpression of the protein and the occurrence of point mutations. Such applications of siRNAs as supplementary therapy could be of clinical significance. The existence of several BCR-ABL fusion transcript variants (eg b3a2 or b2a2) is a major limitation for the application of fusion site-specific siRNAs. However, Wohlbold et al demonstrated in a follow-up study that all common BCR-ABL transcript variants could be successfully targeted with siRNAs that are homologous to the corresponding fusion sites.
TEL-PDGFβR The chromosomal translocation t(5;12)(q33;p13) associated with chronic myelomonocytic leukemia (CMML) generates the TEL-PDGFβR fusion gene[39]. Similar to BCR-ABL, the TEL-PDGFbR fusion protein exhibits a constitutively active tyrosine kinase activity, which can be inhibited with imatinib mesylate or rapamycin. However, as with BCR-ABL, drug resistance due to point mutations limits the therapeutic efficacy of these kinase inhibitors.
Chen et al applied an oncoretroviral transduction system for the delivery of TEL-PDGFβR shRNAs[40]. The siRNAs were transcribed using an H1-promoter-based short hairpin RNA expression system in a self-inactivating retroviral vector. The TEL-PDGFβR siRNAs potently downmodulated TEL-PDGFβR, which affected signal transduction through PI3 kinase and mammalian target of rapamycin (MTOR), and, consequently, inhibited the proliferation of TEL-PDGFβR transformed cells. Furthermore, TEL-PDGFβR depletion sensitized Ba/F3 cells expressing either wild-type TEL-PDGFβR or a mutated, chemoresistant form to the small molecule inhibitors. Moreover, to evaluate the therapeutic efficacy of the siRNA in vivo, TEL-PDGFβR-transformed Ba/F3 cells with or without coexpression of the active siRNA were injected into the tail vein of nude mice. Whereas Ba/F3 cells stably expressing TEL-PDGFβR alone caused tumor development and death with a median latency of 24 d after injection, expression of TEL-PDGFβR siRNA resulted in significantly prolonged survival with a median latency of 41 d.
NPM-ALK Up to 75% of childhood and adolescent anaplastic large cell lymphomas (ALCL) carry the translocation t(2;5)(p23;q35). In this case, the ubiquitously expressed nucleophosmin (NPM) is fused to the cytoplasmic tail of the anaplastic lymphoma tyrosine kinase (ALK). The NPM-ALK fusion protein codes for a constitutively active tyrosine kinase, which is necessary for cell transformation. This kinase enhances factor-independent proliferation and inhibits apoptosis mostly via PI3K–Akt, Jak3/2-Stat3/5 and phospholipase Cγ signaling pathways. Ritter and collaborators designed and evaluated 3 chemically synthesized siRNAs for downregulation of the NPM-ALK fusion mRNA[41]. The most potent of them reduced the levels of NPM-ALK mRNA expression in SR786 ALCL cells by 50%–60%. However, repeated transfections were needed for significant reductions in the protein level, probably due to the long half-life of the NPM-ALK protein. Nevertheless, the siRNAs in this case were successfully used to dissect the signaling pathways employed by NPM-ALK. Similar to TEL-PDGFβR, stable expression of an NPM-ALK shRNA sensitized NPM-ALK-transformed Ba/F3 cells to rapamycin-induced cell death[40].
FLT3 Due to internal tandem duplication (ITD) within the juxtamembrane domain or due to point mutations, FMS-like tyrosine kinase 3 (FLT3) is constitutively activated in 35% of all AML cases. In ALL carrying mixed lineage leukemia gene (MLL) rearrangements, constitutive FLT3 activation is caused by overexpression of this kinase. Similar to other studies[38], Walters et al examined the effects of siRNA-mediated FLT3 suppression on the sensitivity towards a small-molecule FLT3 inhibitor, MLN518[42]. SiRNA-mediated FLT3 depletion diminished the phosphorylation of several downstream molecules, thereby leading to the decreased viability of Ba/F3 and Molm-14 cells, which have internal tandem duplication of FLT3. The combination of FLT3 siRNAs together with the specific FLT3 inhibitor MLN518 led to synergistic effects on cell proliferation and apoptosis induction.
LYN As already described earlier, imatinib mesylate resistance due to BCR-ABL point mutations is a major problem in CML therapy. Therefore, new, complementary approaches to interfere with BCR-ABL function such as siRNAs are currently being intensively studied. Alternatively, proteins other than BCR-ABL, which are also crucial for maintaining the leukemic phenotype, may be promising targets. Members of the SRC kinase family such as SRC or LYN play a central role in BCR-ABL-associated leukemogenesis. For that reason, Ptasznik et al studied the effects of LYN depletion on the survival of imatinib mesylate-resistant CML and ALL blast crisis cells[43]. LYN siRNA reduced LYN protein in both normal hemopoietic cells and BCR-ABL-expressing blasts by 80%–95%. Within 48 h, both siRNA-treated CML and ALL blasts underwent apoptosis, whereas normal cells remained viable. Notably, ALL blasts seemed to be more affected by LYN depletion than CML blasts. This increased dependence of BCR-ABL-positive leukemic blasts on LYN signaling provides the rationale for a complementary treatment of imatinib mesylate-resistant CML blast crisis, particularly when lymphoid in nature.
Targeting transcriptional modulators with siRNAs
AML1/MTG8 The chromosomal translocation t (8;21)(q22;q22), which is the most frequent aberration associated with 10%–15% of all cases of AML, fuses the DNA-binding domain of the transcription factor RUNX1 (also called AML1 or CBFa) to the almost complete open reading frame of RUNXT1 (also named MTG8, ETO or CBFA2T1)[44]. Whereas RUNX1 is an essential transcription factor for definitive hemopoiesis, RUNXT1 or MTG8 is part of histone deacetylase-containing complexes. Thus, the translocation converts a transcriptional modulator to a constitutive repressor of gene expression. The resulting fusion protein AML1/MTG8 (AML1-ETO) inhibits myelopoiesis and supports the clonal expansion of hemopoietic stem cells. Moreover, by directly binding to and sequestering transcription factors, such as SMAD3, C/EBPα or vitamin D receptor, AML1/MTG8 interferes with signal transduction pathways controlling differentiation and proliferation[45].
We examined the efficacy and specificity of siRNAs homologous to the fusion site of AML1/MTG8 transcripts. Such siRNAs efficiently and specifically suppressed AML1/MTG8 without interfering with RUNX1 expression. AML1/MTG8 depletion led to severely impaired clonogenicity, a senescence-associated G1 cell cycle arrest, and an increased responsiveness to Tumor Growth Factor-β (TGFβ)/vitamin D3-induced myelo-monocytic differentiation in t(8;21)-positive leukemic cells[46]. The data imply a central role of AML1/MTG8 not only in the expansion of preleukemic progenitor cells, but also in the maintenance of the leukemia. Moreover, AML1/MTG8 siRNAs are not only of value for the functional analysis of this fusion gene, but may also be useful for an antileukemic therapy.
MLL-AF4 The chromosomal translocations involving human mixed lineage leukemia gene MLL located on chromosome 11 are associated with aggressive lymphoid and myeloid leukemias[47]. MLL is the human trithorax homologue and stabilizes unmethylated CpG islands. It is part of a several MDa large protein complex involved in the regulation of DNA methylation and histone acetylation and methylation. MLL can be fused to more than 40 different partner genes, yielding a very diverse collection of chimeric fusion proteins[48]. The translocation partners do not share any unifying properties. Nevertheless, 2 different classes can be distinguished. One group of partners such as AF1 or GAS7 cause oligomerization of the corresponding fusion protein[49], whereas a second group (eg AF4, AF9, ENL) might be part of a single multiprotein complex[50,51]. The molecular mechanisms of MLL-associated leukemogenesis are currently being intensively studied. Despite the diversity of the fusion proteins, characteristic features of MLL-associated leukemias are an inappropriate expression of a subset of homeotic genes, such as MEIS1 and HOXA9, and the overexpression of FMS-like tyrosine kinase 3 (FLT3)[52].
The translocation t(4;11) is associated with a very aggres-sive and therapy-resistant acute lymphoblastic leukemia in infants and with therapy-related secondary leukemias[53,54]. Recently, we showed that transient inhibition of MLL-AF4 expression with small interfering RNAs impaired the proliferation and clonogenicity of the t(4;11)-positive human leukemic cell lines SEM and RS4;11[19]. Reduction of MLL-AF4 levels induced apoptosis associated with caspase-3 activation and diminished BCL-XL expression. Suppression of MLL-AF4 was paralleled by a decreased expression of the homeotic genes HOXA7, HOXA9, and MEIS1. Moreover, MLL-AF4 depletion inhibited expression of the hemopoietic stem cell marker CD133, indicating hemopoietic differentiation. Finally, transfection of leukemic cells with MLL-AF4 siRNAs reduced leukemia-associated morbidity and mortality in xenotran-splanted SCID mice, suggesting that MLL-AF4 depletion negatively affects leukemia-initiating cells. Our findings demonstrate that MLL-AF4 is important for leukemic clonogenicity and engraftment of this highly aggressive leukemia.
To date, the presence of MLL rearrangements are indicative of a poor clinical outcome, partially due to the lack of specific pharmacological inhibitors[55]. Therefore, the siRNA approach might be an option for developing new strategies for the treatment of these aggressive leukemias.
Challenges and promises
The examples discussed in the previous sections strongly suggest that siRNA-mediated oncogene suppression may become a promising option in antileukemic therapy. However, there are major obstacles to overcome, such as specificity, induction of an interferon response, emergence of escape mutations, or inefficient systemic siRNA delivery in vivo. Only if these challenges can be successfully addressed will siRNA technology fulfill its promises in cancer therapy.
siRNA specificity and possible off-target effects should be given particular consideration. Like all other antisense molecules, siRNAs tolerate mismatches to a certain extent, possibly compromising their sequence specificity[56]. Even if mismatches prevent RISC-mediated cleavage of unintended target transcripts, protein expression may still be affected[57]. Furthermore, siRNAs or shRNAs may compete with endogenously expressed miRNAs for RISC components, thereby affecting miRNA-regulated gene expression and cellular processes such as differentiation[58,59]. Finally, both chemically synthesized siRNAs as well as intracellularly expressed shRNAs may induce a limited interferon response both in cell culture and in vivo[60]. This induction proceeds via Toll-like receptors and/or protein kinase R, but the parameters responsible are just currently becoming clear[61-63]. One possible method of controlling siRNA- triggered interferon response is to examine the induction of classical interferon response genes such as STAT1 and OAS1. Furthermore, ectopic expression of an siRNA-insensitive variant of the target protein would be a suitable control for the possible side-effects of siRNAs. However, such rescue experiments are sometimes difficult to perform. For instance, ectopic expression of AML1/MTG8 or MLL-AF4 inhibits cell proliferation and induces cell death in many different cell types. An alternative might be the sequence-specific inhibition of different variants of the fusion gene. For instance, we targeted 2 different MLL-AF4 variants with specific siRNAs[19]. Each siRNA only inhibited the proliferation of that cell line expressing the perfectly complementary variant, but had no effect on the other cell line. Because both siRNAs are functional in their corresponding cell lines, such an experimental setting is equivalent to a rescue experiment.
Another point of concern is the ability of siRNAs to remain functional in the context of possible escape mutations. However, we and others have demonstrated that different fusion gene variants can be successfully targeted with siRNAs[19,38]. Thus, it may be possible to counteract an escape mutation by simply adapting the siRNA sequence.
The most challenging problem for the therapeutic application of siRNAs is the efficient delivery of siRNA into leukemic tissues, including leukemic stem cells. To date, several techniques have been examined to obtain systemic siRNA delivery in mouse models[64]. However, approaches such as high-pressure, high-volume intravenous injection of synthetic siRNAs, the so-called hydrodynamic delivery, are of limited clinical use because of the severe side effects[65].
Recently, the pharmacokinetics and efficacies of chemically modified siRNAs containing a 5'-cholesterol moiety on the sense strand in combination with limited phosphoro-thioate and 2'-methoxy modification were examined in a mouse model. Intravenous injection resulted in the silencing of ApoB mRNA in liver and jejunum, decreased plasma levels of ApoB protein, and reduced total cholesterol[66]. Alterna-tively, unmodified siRNAs have been successfully delivered by using vehicles such as polyethylenimine, atelocollagen, cationic lipids or neutral liposomes[67–73]. In all these cases, intravenous injection of the corresponding vehicle-siRNA mixture inhibited gene expression and tumor growth in mouse models.
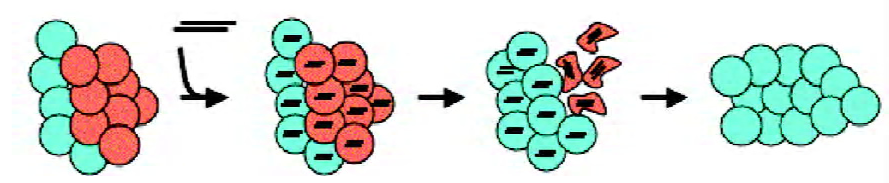
The major goal for RNAi approaches for cancer therapy is the selective elimination of tumor cells without damaging normal cells. For that, if targeting a non-mutated gene, siRNAs should be selectively delivered to the transformed cells. Alternatively, siRNAs may target genes that are exclusively expressed in cancer cells, and that are crucially involved their growth or survival (Figure 3). The latter option is preferable when a fusion gene is involved in tumorigenesis. The examples discussed in this review demonstrate the suitability and effectiveness of this approach both in cell culture and, after siRNA delivery ex vivo, also in murine model systems. Given the rapid progress being made in the field of therapeutic RNA interference, antileukemic siRNAs may be not too far away from complementing existing therapeutic protocols to treat leukemia.
References
- Novina CD, Sharp PA. The RNAi revolution. Nature 2004;430:161-4.
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-11.
- Huppi K, Martin SE, Caplen NJ. Defining and assaying RNAi in mammalian cells. Mol Cell 2005;17:1-10.
- Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev 1999;13:3191-7.
- Caplen NJ. Gene therapy progress and prospects. Downregulating gene expression: the impact of RNA interference. Gene Ther 2004;11:1241-8.
- Cheng JC, Moore TB, Sakamoto KM. RNA interference and human disease. Mol Genet Metab 2003;80:121-8.
- Borkhardt A, Heidenreich O. RNA interference as a potential tool in the treatment of leukaemia. Expert Opin Biol Ther 2004;4:1921-9.
- Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med 2005;11:71-6.
- Claus R, Almstedt M, Lubbert M. Epigenetic treatment of hematopoietic malignancies: in vivo targets of demethylating agents. Semin Oncol 2005;32:511-20.
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev 2001;15:188-200.
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005;436:740-4.
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 2004;15:185-97.
- Okamura K, Ishizuka A, Siomi H, Siomi MC. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev 2004;18:1655-6.
- Ambros V. The functions of animal microRNAs. Nature 2004;431:350-5.
- Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002;297:2056-60.
- Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA 2002;8:855-60.
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 2005;7:719-23.
- Sen GL, Blau HM. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol 2005;7:633-6.
- Thomas M, Gessner A, Vornlocher HP, Hadwiger P, Greil J, Heidenreich O. Targeting MLL-AF4 with short interfering RNAs inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood 2005;106:3559-66.
- Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A, et al. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res 2004;32:936-48.
- Fedorov Y, King A, Anderson E, Karpilow J, Ilsley D, Marshall W, et al. Different delivery methods-different expression profiles. Nat Methods 2005;2:241.
- Donze O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res 2002;30:e46.
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002;2:243-7.
- Abbas-Terki T, Blanco-Bose W, Deglon N, Pralong W, Aebischer P. Lentiviral-mediated RNA interference. Hum Gene Ther 2002;13:2197-201.
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
- Barthe C, Gharbi MJ, Lagarde V, Chollet C, Cony-Makhoul P, Reiffers J, et al. Mutation in the ATP-binding site of BCR-ABL in a patient with chronic myeloid leukaemia with increasing resistance to STI571. Br J Haematol 2002;119:109-11.
- Barthe C, Cony-Makhoul P, Melo JV, Mahon JR. Roots of clinical resistance to STI-571 cancer therapy. Science 2001;293:2163.
- Skorski T, Nieborowska-Skorska M, Nicolaides NC, Szczylik C, Iversen P, Iozzo RV, et al. Suppression of Philadelphia1 leukemia cell growth in mice by BCR-ABL antisense oligodeoxynucleo-tide. Proc Natl Acad Sci USA 1994;91:4504-8.
- Wilda M, Fuchs U, Wossmann W, Borkhardt A. Killing of leukemic cells with a BCR/ABL fusion gene by RNA interference (RNAi). Oncogene 2002;21:5716-24.
- Scherr M, Battmer K, Winkler T, Heidenreich O, Ganser A, Eder M. Specific inhibition of bcr-abl gene expression by small interfering RNA. Blood 2003;101:1566-9.
- Rapozzi V, Xodo LE. Efficient silencing of bcr/abl oncogene by single- and double-stranded siRNAs targeted against b2a2 transcripts. Biochemistry 2004;43:16134-41.
- Li MJ, McMahon R, Snyder DS, Yee JK, Rossi JJ. Specific killing of Ph+ chronic myeloid leukemia cells by a lentiviral vector-delivered anti-bcr/abl small hairpin RNA. Oligonucleotides 2003;13:401-9.
- Scherr M, Battmer K, Schultheis B, Ganser A, Eder M. Stable RNA interference (RNAi) as an option for anti-bcr-abl therapy. Gene Ther 2005;12:12-21.
- Withey JM, Marley SB, Kaeda J, Harvey AJ, Crompton MR, Gordon MY. Targeting primary human leukaemia cells with RNA interference: Bcr-Abl targeting inhibits myeloid progenitor self-renewal in chronic myeloid leukaemia cells. Br J Haematol 2005;129:377-80.
- Zhelev Z, Bakalova R, Ohba H, Ewis A, Ishikawa M, Shinohara Y, et al. Suppression of bcr-abl synthesis by siRNAs or tyrosine kinase activity by Glivec alters different oncogenes, apoptotic/antiapoptotic genes and cell proliferation factors (microarray study). FEBS Lett 2004;570:195-204.
- Ohba H, Zhelev Z, Bakalova R, Ewis A, Omori T, Ishikawa M, et al. Inhibition of bcr-abl and/or c-abl gene expression by small interfering, double-stranded RNAs: cross-talk with cell proliferation factors and other oncogenes. Cancer 2004;101:1390-403.
- Bartova E, Harnicarova A, Pachernik J, Kozubek S. Nuclear topography and expression of the BCR/ABL fusion gene and its protein level influenced by cell differentiation and RNA interference. Leuk Res 2005;29:901-13.
- Wohlbold L, van der Kuip H, Miething C, Vornlocher HP, Knabbe C, Duyster J, et al. Inhibition of bcr-abl gene expression by small interfering RNA sensitizes for imatinib mesylate (STI571). Blood 2003;102:2236-9.
- Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomono-cytic leukemia with t(5;12) chromosomal translocation. Cell 1994;77:307-16.
- Chen J, Wall NR, Kocher K, Duclos N, Fabbro D, Neuberg D, et al. Stable expression of small interfering RNA sensitizes TEL-PDGFbetaR to inhibition with imatinib or rapamycin. J Clin Invest 2004;113:1784-91.
- Ritter U, Damm-Welk C, Fuchs U, Bohle RM, Borkhardt A, Woessmann W. Design and evaluation of chemically synthesized siRNA targeting the NPM-ALK fusion site in anaplastic large cell lymphoma (ALCL). Oligonucleotides 2003;13:365-73.
- Walters DK, Stoffregen EP, Heinrich MC, Deininger MW, Druker BJ. RNAi-induced down-regulation of FLT3 expression in AML cell lines increases sensitivity to MLN518. Blood 2005;105:2952-4.
- Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat Med 2004;10:1187-9.
- Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol 1999;106:296-308.
- Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol 1998;18:322-33.
- Heidenreich O, Krauter J, Riehle H, Hadwiger P, John M, Heil G, et al. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood 2003;101:3157-63.
- Pui CH, Schrappe M, Ribeiro RC, Niemeyer CM. Childhood and adolescent lymphoid and myeloid leukemia. Hematology (Am Soc Hematol Educ Program) 2004.118-45.
- Rowley JD. The role of chromosome translocations in leukemogenesis. Semin Hematol 1999;36:59-72.
- So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell 2003;4:99-110.
- Srinivasan RS, Nesbit JB, Marrero L, Erfurth F, LaRussa VF, Hemenway CS. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia 2004;18:1364-72.
- Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 2005;24:5525-32.
- Armstrong SA, Golub TR, Korsmeyer SJ. MLL-rearranged leukemias: insights from gene expression profiling. Semin Hematol 2003;40:268-73.
- Domer PH, Fakharzadeh SS, Chen CS, Jockel J, Johansen L, Silverman GA, et al. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci USA 1993;90:7884-8.
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41-7.
- Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med 2004;350:1535-48.
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J 2001;20:6877-88.
- Saxena S, Jonsson ZO, Dutta A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem 2003;278:44312-9.
- Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004;303:83-6.
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell 2005;120:635-47.
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 2003;34:263-4.
- Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 2004;101:1892-7.
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 2005;11:263-70.
- Kim DH, Longo M, Han Y, Lundberg P, Cantin E, Rossi JJ. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat Biotechnol 2004;22:321-5.
- Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med 2003;9:347-51.
- Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004;432:173-8.
- Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res 2004;32:e149.
- Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther 2005;12:461-6.
- Takeshita F, Minakuchi Y, Nagahara S, Honma K, Sasaki H, Hirai K, et al. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci USA 2005;102:12177-82.
- Yano J, Hirabayashi K, Nakagawa S, Yamaguchi T, Nogawa M, Kashimori I, et al. Antitumor activity of small interfering RNA/cationic liposome complex in mouse models of cancer. Clin Cancer Res 2004;10:7721-6.
- Landen CN Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res 2005;65:6910-8.
- Thomas M, Gessner A, Vornlocher HP, Hadwiger P, Greil J, Heidenreich O. Targeting MLL-AF4 with short interfering RNAs inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood 2005;106:3559-66.
- Heidenreich O, Krauter J, Riehle H, Hadwiger P, John M, Heil G, et al. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood 2003;101:3157-63.
- Martinez N, Drescher B, Riehle H, Cullmann C, Vornlocher HP, Ganser A, et al. The oncogenic fusion protein RUNX1-CBFA2T1 supports proliferation and inhibits senescence in t(8;21)-positive leukaemic cells. BMC Cancer 2004;4:44.