
Phosphorylated heat shock protein 27 is involved in enhanced heart tolerance to ischemia in short-term type 1 diabetic rats1
Introduction
It is well known that cardiovascular disease is the major cause of morbidity and mortality in diabetic patients. However, there exists considerable debate over the causal relationship between diabetes per se and cardiovascular disease[1]. Unlike classical microvascular complications, large vessel atherosclerosis is not correlated with duration and severity of hyperglycemia[2,3]. Some studies demonstrate that cardiovascular disease prevalence remains constant despite different levels of glycohemoglobin. Furthermore, intensive control of hyperglycemia is not consistently effective in reducing cardiovascular complications[4], leaving unanswered the question of whether hyperglycemia contri-butes to cardiovascular disease in diabetes.
In addition to clinical debates, experimental studies also show inconsistent results regarding the role of hyperglycemia in myocardial ischemia. Studies of animals with type 1 diabetes demonstrate either increased or decreased sensitivity of diabetic hearts to ischemia[5–7]. Suggested mechanisms for decreased sensitivity include attenuated Na+-H+ exchanger activity, inhibited glycolysis, and reduced Ca2+ overload during reperfusion[7–9]. Studies demonstrating the detrimental effects of hyperglycemia on ischemia suggest roles of pseudohypoxia and reactive oxygen species[6]. Regardless of whether hyperglycemia is the cause of increased cardiovascular disease, it is generally agreed that experimental type 2 diabetes has a detrimental effect on myocardial ischemia[10] and coexisting cardiovascular risk factors like hypertension and elevated cholesterol contribute significantly to the poor outcome in type 2 diabetes as seen in clinical settings[11,12]. Type 1 diabetes, however, has severe hyperglycemia and relatively less coexisting cardiovascular disease. Thus, type 1 diabetic animals provide good models for investigating hyperglycemia influence on ischemic outcomes.
The present study was designed to observe heart sensitivity to ischemia in type 1 diabetic rats using functional recovery and creatine phosphokinase as measures of ischemic injury, in addition to arrhythmias. Aortic vessel function was also examined to test whether there was functional impairment of large vessels. As circulating antibody titers to some of heat shock proteins family are increased in type 1 diabetic patients[13], we observed whether these powerful protective proteins were upregulated in diabetic hearts. Our data indicate that type 1 diabetic hearts are resistant to ischemia/reperfusion with no obvious impairment of large vessel relaxation. The study also reveals increased expression of myocardial phosphorylated heat shock protein 27 (phospho-hsp27) in diabetes. To further examine the association of hyperglycemia and heat shock proteins with heart sensitivity to ischemia, we observed the effects of insulin treatment on ischemic outcomes and phospho-hsp27 by the correction of hyperglycemia.
Materials and methods
Animals Male Sprague-Dawley rats (originally weighing 200–220 g) were injected intraperitoneally with 60 mg/kg of streptozocin (Sigma, St Louis, USA) in citrate buffer (pH 4.5). Age-matched control rats received an injection of citrate buffer. The onset of diabetes was confirmed by measuring blood glucose within 72 h after injection. Rats had free access to tap water and standard laboratory chow and were maintained until the in vitro experiment began after 3 weeks of hyperglycemia induction. Another experiment was carried out with one group of rats treated with subcutaneous injection of protamine zinc insulin (9 units/kg) 3 weeks after diabetes induction and a heart ischemia experiment was carried out after 2 d of complete correction of hyperglycemia (duration of insulin administration was 5.1±0.7 d). All experiments conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publication N
Isolated heart preparation and ischemia/reperfusion The hearts were excised under anesthesia with sodium pentobarbital 60 mg/kg, ip, and perfused retrogradely (Langendorff apparatus, IPH-W2, Labo Support, Osaka, Japan) through the aorta with Krebs-Henseleit buffer (KHB) containing: NaCl 118.0 mmol/L, KCl 4.7 mmol/L, CaCl2 2.5 mmol/L, MgSO4 1.2 mmol/L, KH2PO4 1.2 mmol/L, NaHCO3 25.0 mmol/L, glucose 11.0 mmol/L, Na2-EDTA 0.5 mmol/L. After cannulation of the left atrium the hearts were switched to antegrade perfusion (working heart). Perfusate was kept at 37 ºC and bubbled with 95% O2 and 5% CO2. Aortic perfusion pressure was set at 60 mmHg and the left atrium at 10 mmHg at the beginning of each experiment. All hearts were allowed to beat spontaneously.
With a catheter inserted into the left ventricular cavity, left ventricular pressure (LVP), left ventricular end-diastolic pressure (LVEDP) and maximal rate of LVP rise (dp/dtmax) and fall (dp/dtmin) were recorded with a data acquisition system (MPA 2000, Alcott Biotech, Shanghai, China). The hearts were subjected to 30-min global ischemia by clamping both atrial inflow and aortic outflow and followed by 40 min of reperfusion.
Two silver electrodes were inserted into the myocardium and electrocardiogram was recorded continuously. Premature ventricular beats, ventricular tachycardia and fibrillation during reperfusion were recorded. The severity of arrhythmias was scored on a 0 to 9 scale[14]. The time from the cessation of perfusion to the cessation of heart contraction was recorded as time to arrest.
Creatine phosphokinase and norepinephrine assay Creatine phosphokinase (CPK) was measured in coronary effluent with assay kits (Jian-Cheng Biomedical Engineering, Nanjing, China) according to the manufacturer’s protocols. Coronary norepinephrine was determined with high performance liquid chromatography coupled with electrochemical detection (Neurochem Model 5500, ESA, Bedford, MA, USA).
Vessel tension recording Aortic rings were suspended in an organ bath (PowerLab ML0140, ADInstruments, Castle Hill, NSW, Australia) containing KHB. The buffer was bubbled with 95% O2 and 5% CO2 and kept at 37 ºC. Aortic rings were allowed to equilibrate for 90 min before tension recording. After precontraction with 1×10-7 mol/L of norepinephrine, acetylcholine (ACh) was added in a cumulative manner and aorta relaxation was recorded. Vasodilation in the presence of nitric oxide synthase inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME, 1×10-4 mol/L) was also measured.
Western blot analysis Left ventricles before and after ischemia were freeze-clamped and homogenized in lysis buffer (pH 7.4) containing: Tris/HCl 50 mmol/L, sucrose 150 mmol/L, edetic acid-Na2 5 mmol/L, egtazic acid 2 mmol/L, Na3VO4 1 mmol/L, NaF 50 mmol/L, phenylmethanesulfonyl fluoride 0.1 mmol/L, leupeptin 1 mg/L. Protein samples were loaded onto 12% SDS-PAGE acrylamide gels and transferred to polyvinyli-dene difluoride membrane. Blots were incubated with antibodies to either hsp27 or phosphorylated hsp27 at serine 15 and inducible hsp70 (Santa Cruz, Santa Cruz, CA, USA). The immunoreactive bands were visualized using enhanced chemiluminescence detection, and relative levels of heat shock proteins were semiquantified with densitometry (QuantityOne software, Bio-Rad, Hercues, CA, USA) normalizing to actin band.
Statistical analysis Data were expressed as mean±SEM. Parameters were compared with analysis of variance (ANOVA) for repeated measures followed by Student-Newman-Keuls test. The relationship between blood glucose level and ischemic outcomes (arrhythmia score and time to arrest) was evaluated with multiple regression analysis. P<0.05 was considered statistically significant.
Results
Effects of diabetes on baseline and post-ischemic heart function A significant elevation in blood glucose was accompanied by a reduction in contractility of the diabetic heart, represented by lower dp/dtmax and lower heart work (lower left ventricular developed pressure, LVDP; the difference between LVP and LVEDP). Diabetic hearts also exhibited reduced heart rate and normal LVEDP at baseline perfusion(Table 1).
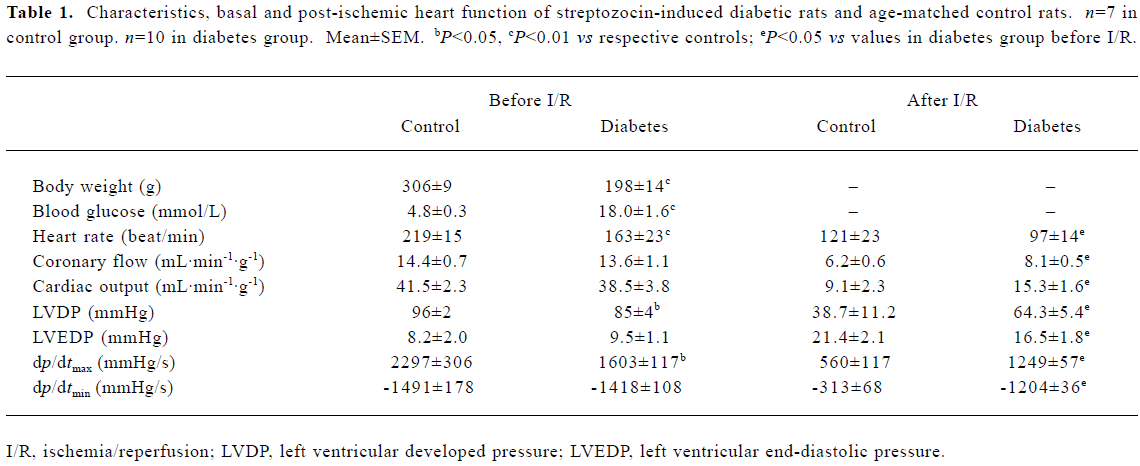
Full table
Although depressed at the baseline, post-ischemic recovery of dp/dtmax, dp/dtmin and LVDP in diabetic hearts was enhanced significantly as compared with controls (Table 1). Coronary perfusion and cardiac output also recovered to better levels after ischemia in diabetes (Table 1). Reperfusion arrhythmias, cardiac CPK release, as well as cardiac norepinephrine release, were reduced in diabetic hearts (Table 2).

Full table
Ischemic outcome and correlation with blood glucose level Subgroups of mild or severe hyperglycemia (blood glucose <19 or >20 mmol/L) exhibited different post-ischemic outcomes (Figure 1A). Higher glucose within each group was accompanied by longer time to arrest and less arrhythmias (Figure 1B, 1C).

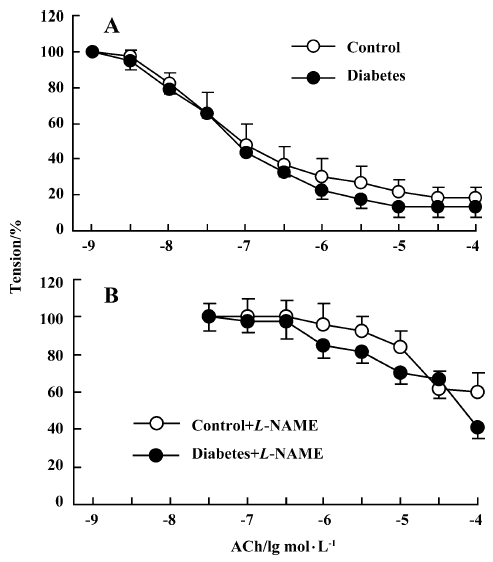
Aortic relaxation Normal vessel relaxation was found in the diabetic aorta (Figure 2), indicating no obvious impairment of large vessel function in this type of diabetic rat. The ACh-induced relaxation was abrogated by 1×10-4 mol/L L-NAME to similar extents in 2 groups.
Myocardial expression of heat shock proteins There were no differences in basal amounts of total hsp27 or inducible hsp70 between diabetic and control groups (Figure 3A, 3B). Because the protective ability of hsp27 depends on its phosphorylation state, we also analyzed phosphorylated hsp27 in the myocardium. Phospho-hsp27 content was found to be significantly higher in diabetic hearts before (Figure 3C) and after ischemia (Figure 3D). To eliminate the possible role of streptozocin in inducing expression of phospho-hsp27, we also examined the content of phospho-hsp27 in rats that failed to develop diabetes after streptozocin injection (15% of animals receiving streptozocin did not show significant elevation of blood glucose in this study and served as non-diabetic controls). There was no significant change of phospho-hsp27 in those animals (relative density values for myocardial phospho-hsp27 before ischemia were 5.4±0.9 in normal controls vs 6.2±1.0 in non-diabetic controls receiving streptozocin, n=4, P>0.05).

Ischemic outcomes after metabolic correction Further intervention experiments were carried out with complete correction of hyperglycemia for 2 d prior to the ischemia experiment (Figure 4A). Cardioprotection was abrogated in blood glucose-controlled diabetes, demonstrated by increased reperfusion arrhythmias (Figure 4B) and reduced post-ischemic dp/dtmax (Figure 4C). Abrogation of cardioprotection through metabolic control was accompanied by reduction of myocardial phospho-hsp27 (Figure 4D).

Discussion
The present study demonstrated significantly enhanced resistance to ischemia/reperfusion in type 1 diabetic hearts, manifested by improved post-ischemic mechanical function, prolonged time to arrest, attenuated reperfusion arrhythmias and reduced CPK release. Furthermore, there was a significantly increased amount of phospho-hsp27 in diabetic hearts compared with non-diabetic hearts. The reduction of phospho-hsp27 in correlation to hyperglycemia was effective in abrogating heart tolerance to ischemia in diabetes.
Although there is controversy over the effects of hyperglycemia on cardiovascular outcomes, it is important to note that increased tolerance to ischemia is almost exclusively found in type 1 diabetic hearts[8,9,15,16]. In contrast to decreased sensitivity to ischemia of type 1 diabetic hearts, increased sensitivity to myocardial ischemia was found in type 2 diabetic Otsuka Long-Evans Tokushima Fatty rats[17]. It is of interest that higher blood glucose induced by sucrose feeding in type 2 diabetes paradoxically attenuated post-ischemic cardiac dysfunction[17]. There is also a study showing that the correction of metabolic state alone in type 2 diabetes did not improve heart function recovery from ischemia[10]. These data from type 1 and type 2 diabetes indicate that factors other than hyperglycemia increase heart sensitivity to ischemia in type 2 diabetes, and marked high glucose or other factors secondary to elevated glucose can participate in preconditioning hearts to resist ischemia insult in type 1 diabetes.
A striking finding of the present study is that increased myocardial resistance to ischemia was accompanied by increased amounts of phospho-hsp27 in diabetic hearts. Because there is no significant change in myocardial phospho-hsp27 of rats that failed to develop diabetes after injection of streptozocin, the increase of phospho-hsp27 in streptozocin-induced diabetes is not likely to be induced by this toxic agent used for destroying pancreas beta-cells and induction of type 1 diabetes. Here we suggest for the first time that hyperglycemia can present a chronic and mild, yet stressful, stimulus to myocardium inducing upregulation of some stress proteins in diabetic hearts, which may in turn enhance heart tolerance to ischemia.
Elevated levels of hsp27 are reported to participate in cardioprotection by maintaining the integrity of microtubules and actin cytoskeleton, and can protect endothelium from ischemia[18]. The phosphorylation and translocation of hsp27 from cytosol to myofibril or nucleus is found to be especially important for protecting actin fragmentation and microtubule degradation[19]. In the present study, hyperglycemia did not affect total hsp27 but significantly increased the amount of phosphor-hsp27, indicating the potential cardioprotective role of phospho-hsp27 during ischemia and reperfusion of diabetic hearts. The elevated lipid peroxidation product found in our previous work and other works[6,20] indicates higher oxidative stress in type 1 diabetes, while upregulation of protective proteins may compensate for such detrimental effects of hyperglycemia.
Inhibited glycolysis could result in myocardial salvage as a result of less accumulation of lactate during ischemia[21]. In accordance with previous published reports, we also found decreased lactate production from diabetic hearts[20], which possibly contributed to the observed cardioprotection. In addition to the above mechanisms, decreased cardiac norepinephrine may also account for improved mechanical function, in accordance with our previous finding that norepinephrine reduction conferred significant cardioprotection during ischemia[22]. Because Na+-H+ exchanger enhances norepinephrine release, diminished activation of Na+-H+ exchanger in diabetic hearts[8] may be one explanation for reduced norepinephrine release. This needs further investi-gation.
There is a report that early type 1 diabetes was found to have increased aortic relaxation[23]. In this study, no significant large vessel dysfunction was found in this early stage of type 1 diabetes. The relatively higher post-ischemic coronary flow in the diabetic group also indicated no impairment of coronary vessel function. The elevated coronary perfusion may be a result of myogenic dilation, a main determinant for myocardial perfusion, as during reperfusion the diabetic hearts relaxed significantly more than the controls. The improved coronary perfusion in turn enhanced myocardial salvage during reperfusion.
In summary, the present study reveals that the hearts of early-stage type 1 diabetes are resistant to ischemic injury. The concomitant upregulation of phospho-hsp27, in addition to inhibited glycolysis and reduced norepinephrine release, may participate in protecting diabetic hearts from ischemia. Hyperglycemia may contribute to the induction of the stress protein. However, caution should be taken when extrapolating the present results to clinical settings as most diabetic patients have type 2 diabetes commonly associated with other risk factors. Further investigation on the combination of hyperglycemia and other conventional risk factors will lead to a better understanding of the complex pathogenesis of diabetes mellitus.
References
- Buse JB. Should postprandial glucose be routinely measured and treated to a particular target? No! Diabetes Care 2003;26:1615-8.
- Balkau B, Forhan A, Eschwege E. Two hour plasma glucose is not unequivocally predictive for early death in men with impaired fasting glucose: more results from the Paris Prospective Study. Diabetologia 2002;45:1224-30.
- Jarrett RJ. Type 2 (non-insulin-dependent) diabetes mellitus and coronary heart disease – chicken, egg or neither? Diabetologia 1984;26:99-102.
- Meigs JB, Singer DE, Sullivan LM, Dukes KA, D’Agostino RB, Nathan DM, et al. Metabolic control and prevalent cardiovascular disease in non-insulin-dependent diabetes mellitus (NIDDM): The NIDDM Patient Outcome Research Team. Am J Med 1997;102:38-47.
- Liu Y, Thornton JD, Cohen MV, Downey JM, Schaffer SW. Streptozotocin-induced non-insulin-dependent diabetes protects the heart from infarction. Circulation 1993;88:1273-8.
- Marfella R, D’Amico M, Di Filippo C, Piegari E, Nappo F, Esposito K, et al. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia 2002;45:1172-81.
- Feuvray D, Lopaschuk GD. Controversies on the sensitivity of the diabetic heart to ischemic injury: the sensitivity of the diabetic heart to ischemic injury is decreased. Cardiovasc Res 1997;34:113-20.
- Ramasamy R, Schaefer S. Inhibition of Na+-H+ exchanger protects diabetic and non-diabetic hearts from ischemic injury: insight into altered susceptibility of diabetic hearts to ischemic injury. J Mol Cell Cardiol 1999;31:785-97.
- Tani M, Neely JR. Hearts from diabetic rats are more resistant to in vitro ischemia: possible role of altered Ca2+ metabolism. Circ Res 1988;62:931-40.
- Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 2003;52:434-41.
- Stern MP. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes 1995;44:369-74.
- Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001;37:1053-9.
- Weitgasser R, Lechleitner M, Koch T, Galvan G, Muhlmann J, Steiner K, et al. Antibodies to heat shock protein 65 and neopterin levels in patients with type 1 diabetes mellitus. Exp Clin Endocrinol Diabetes 2003;111:127-31.
- Curtis MJ, Walker MJ. Quantification of arrhythmia using scoring systems: an examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc Res 1988;22:656-65.
- Ravingerova T, Neckar J, Kolar F, Stetka R, Volkovova K, Ziegelhoffer A, et al. Ventricular arrhythmias following coronary artery occlusion in rats: is the diabetic heart less or more sensitive to ischaemia? Basic Res Cardiol 2001;96:160-8.
- Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 1997;46:292-300.
- Chen H, Higashino H, Kamenov ZA, Azuma M, Lee WH, Yang XQ, et al. Preserved postischemic heart function in sucrose-fed type 2 diabetic OLETF rats. Life Sci 2003;72:2839-51.
- Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res 2001;51:637-46.
- Dana A, Skarli M, Papakrivopoulou J, Yellon DM. Adenosine A1 receptor induced delayed preconditioning in rabbits: induction of p38 mitogen-activated protein kinase activation and Hsp27 phosphorylation via a tyrosine kinase- and protein kinase C-dependent mechanism. Circ Res 2000;86:989-97.
- Wang LP, Liu T, Chen H, Chen HZ, Gao PJ, Zhu DL. Experimental study on paradoxical phenomenon in type 1 diabetic heart: increased resistance to ischemia/reperfusion injury. Shanghai Med J 2004;91:300-6.
- Finegan BA, Lopaschuk GD, Gandhi M, Clanachan AS. Ischemic preconditioning inhibits glycolysis and proton production in isolated working rat hearts. Am J Physiol 1995;269:H1767-H1775.
- Chen H, Higashino H, Maeda K, Zhang Z, Ohta Y, Wang Z, et al. Reduction of cardiac norepinephrine improves postischemic heart function in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 2001;38:821-32.
- Shen B, Ye CL, Ye KH, Liu JJ, Sun P, Jiang JH. Mechanism underlying enhanced endothelium-dependent vasodilation in thoracic aorta of early stage streptozotocin-induced diabetic mice. Acta Pharmacol Sin 2003;24:422-8.