
Effects of melatonin on wortmannin-induced tau hyperphosphorylation1
Introduction
Alzheimer disease (AD) is neuropathologically characterized by cell loss and formation of amyloid plaques and neurofibrillary tangles (NFTs), which are composed of paired helical filaments formed by abnormally hyperphosphorylated microtubule associated protein tau[1]. Studies have shown that tau regulates microtubule dynamics, axonal transport, and neuronal morphology by binding and stabilizing the microtubule structure[2,3]. In AD, abnormally phosphorylated tau associated with paired helical filaments decreases affinity in binding to the microtubules and balks its normal microtubule-related function[4]. Therefore, understanding the regulatory mechanism for tau hyperphosphorylation and aggregation is critical in designing the strategies to arrest it.
A defect in the regulation of protein kinases/protein phosphatases is responsible for tau hyperphosphorylation. Among them, glycogen synthase kinase-3 (GSK-3) is a recognized tau kinase that plays a crucial role in AD pathology[5]. GSK-3 phosphorylates tau both in vivo and in intact cells, and enhanced expression of GSK-3 leads to tau hyperphos-phorylation[6,7]. In an Alzheimer's-affected brain, GSK-3 is activated in pretangle neurons and accumulates in paired helical filaments[8]. We also found that the activation of GSK-3 by brain injection of wortmannin led to hyperphosphoryla-tion of tau and spatial memory impairment in a rat brain[9]. These data together indicate that GSK-3 can serve as a crucial target for reproducing an AD-like experimental modal and for screening potential therapeutic reagents. However, it is not known whether wortmannin also induces tau hyperphosphorylation in neuroblastoma N2a cells, and whether wortmannin treatment affects cell viability, what kind of cell death wortmannin might induce, and if melatonin, as an effective antioxidant secreted from pineal gland [10], which has been used in our previous studies[11-13], influences wortmannin-induced alterations in N2a cell and by which mechanism melatonin works.
Therefore, in the present study, we examined the effect of wortmannin, an indirect GSK-3 activator, on cell viability and tau phosphorylation, and the effect and mechanism of melatonin on wortmannin-induced cytotoxicities in N2a cells. We found that the treatment of cells with 1 µmol/L wortmannin induced GSK-3 activation and tau hyperphosphorylation. Although reduced cell viability and increased cell retraction were observed, no obvious cell apoptosis was detected. Melatonin not only attenuated wortmannin-induced lipid peroxidation but also counteracted wortmannin-induced GSK-3 overactivation and tau hyperphosphorylation.
Materials and methods
Measurement of cell viability and metabolism N2a cell, a gift from Dr HX XU (The Burnham Institute, San Diego, USA), was propagated in Dulbecco's modified Eagle's medium with 5% fetal bovine serum (5% CO2 and 95% air) (GIBCO, NY, USA). The cells were then grown and differentiated in 96-well culture plates at density of 1.5×105 cells in 100 µL. Cells were exposed to various concentrations of wortmannin for 2 h at 37 ºC in the presence or absence of a 12-h-preincubation with melatonin 25, 50, and 100 µmol/L or Vitamin E (VE). Dimethylsulfoxide (Me2SO, 0.01%), in which wortmannin, melatonin and VE were dissolved, served as a vehicle control. Then, 0.2% crystal violet or 1% MTT (3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) in phosphate-buffered saline (PBS) were added and incubated for 3 min or 4 h at dark and read at 570 nm (TECAN Austria, Salzburg, Austria). All assays were performed with five repeats. Wortmannin, Me2SO and melatonin were purchased from Sigma (St Louis, MO, USA).
Morphology features of N2a cells with phase-contrast and electron microscopy Morphology changes of N2a cells treated by wortmannin with or without preincubation of melatonin or VE were examined under phase-contrast micro-scope. For electron microscopy, cultures were fixed in 2.5% glutaraldehyde/0.2% tannic acid in 0.1 mol/L cacodylate buffer (pH 7.4) overnight at 4 ºC and then post-fixed in 1% osmium tetroxide/1.5% ferrocyanide solution for 30 min at room temperature. Cells were dehydrated in ethanol, embedded in Epon resin, and heat polymerized. Epon blocks were cut, double stained with uranyl acetate and lead citrate, and observed under an Opton EN/l0C electron microscope (Opton, Oberkochen, Germany).
TUNEL assay and cleavage of caspases-3 To determine the form of cell death in wortmannin-treated cells and relationship of tau phosphorylation and apoptosis, we used the Dead End TM Colorimetric Apoptosis detection system (TUNEL) (Promega Corp, Madison, USA) and immunofluorescence of cleaved caspase-3 to measure DNA fragmentation and caspase-3 activation. Positive control of apoptosis was treated with 1 µmol/L staurosporine, an apoptosis inducer for 3 h. For TUNEL, cells were fixed in 4% paraformaldehyde solution for 25 min at room temperature, rinsed in phosphate buffered saline (PBS), and permeabilized by immersing the slides in 0.2% Triton X-100 solution. Cells were incubated with terminal deoxynucleotidyl transferase (TdT) reaction mixture containing biotinylated nucleotides and TdT at 37 ºC for 60 min, rinsed with SSC (Sodium Chloride-Sodium Citrate Buffer) and PBS. Streptavidin HRP (Horseradish Peroxidase) was added to cells. Slides were then stained with diaminobenzidine system (DAB).
For caspases-3 immunofluorescence assay, cells were fixed for 15 min with 4% paraformaldehyde in 0.1 mol/L phosphate buffer and immunostained by sequential reaction with a rabbit polyclonal antibody caspase-3 (1:200) (Cell Signaling Technology Inc, Beverly, MA, USA), followed by FITC (Fluorescein isothiocyanate)-conjugated goat anti-rabbit IgG (Dianova GmbH, Hamburg, Germany). Sections were analyzed with a 525-nm filter (FITC staining) with a fluorescence microscope (Olympus, Hamburg, Germany).
Western blot Total cell lysates of N2a from each treatment were separated on 10% SDS-PAGE (PolyAcrylamide Gel Electrophoresis) and the protein were transferred onto the nitrocellulose membranes. Membranes were blocked in Tris-buffered saline containing 5% non-fat milk followed by incubation with primary antibody at 4 ºC for 24 h. Primary mouse monoclonal antibodies including Tau-1 and PHF-1 and rabbit polyclonal antibodies 92e which recognized phosphorylated tau and non-phosphorylated tau were gifts from Dr BINDLE (North Western University, Chicago, Illinois), Dr DAVIES (Albert Einstein College of Medicine, Bronx, NY), and Drs Iqbal and Grundke-Iqbal (NYS Institute for Basic Research, Staten Island, NY, USA). GSK3β and phospho-GSK3β (serine9) were obtained from Cell Signaling Technology Inc. (Beverly, MA, USA). The blots were developed by enhanced chemiluminescence (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and computer image analysis software (Image-Pro Plus, Media Cybernetics, Silver Spring, MD) was used for quantitative analysis of the blots.
Kinase assay Activity of GSK-3 was measured using phospho-GS (Upstate Biotechnology, Lake Placid, NY, USA) as substrate. Cells were treated with factors as described earlier and lysed at 4 ºC in a solution containing (in mmol/L) Tris-HCl 10 (pH 7.4), edetic acid 10, NaCl 0.15, NaF 50, Na3VO4 0.2, 1% NP40, phenylmethylsulfonyl fluoride 1, and 2 µg/µL aprotinin. Cell lysates were clarified by centrifugation at 15 000×g at 4 ºC for 10 min. The samples were incubated with the substrates in buffer containing 30 mmol/L Tris-HCl (pH 7.4), 10 mmol/L MgCl2, 10 mmol/L NaF, 1 mmol/L Na3VO4, 2 mmol/L Na4EGTA, 10 mmol/L β-mercaptoethanol, 0.2 mmol/L (1500 cpm/pmol) [γ-32P] ATP (Beijing Yahui Biologic and Medicinal Engineering Co, Beijing, China) at 30 ºC for 30 min. Reaction was stopped by adding 12.5 µL o-phosphoric acid (300 mmol/L). A total of 20 µL of incubation mixture was applied in duplicates to phosphor cellulose units. The filters were washed 3 times with 75 mmol/L o-phosphoric acid and then dried. The radioactivity incorporated into the substrates was analyzed by liquid scintillation counting at 30 ºC. The protein concentration of the cell lyses was estimated by bicinchoninic acid (BCA) method (Micro BCA Protein Assay Reagent Kit, Pierce Biotechnology, Rockford, IL, USA). GSK-3 activity was expressed as nmol·min-1·g-1 protein at 30 ºC.
Assay of malondialdehyde (MDA) and superoxide dismutase (SOD) The amount of MDA was measured by the thiobarbituric acid (TBA) assay, which is based on the reaction of MDA with TBA to produce a red species with excitation at 515 nm and emission at 553 nm measured in Schimadzu-RF-5000 (Kyoto, Japan)[14]. The activity of SOD was measured according to the method established previously[15].
Data analysis Data were expressed as mean±SD and analyzed using SPSS 10.0 statistical software (SPSS Inc, Chicago, Illinois, USA). The one-way ANOVA procedure followed by Student's t-test was used to determine the differences among groups.
Results
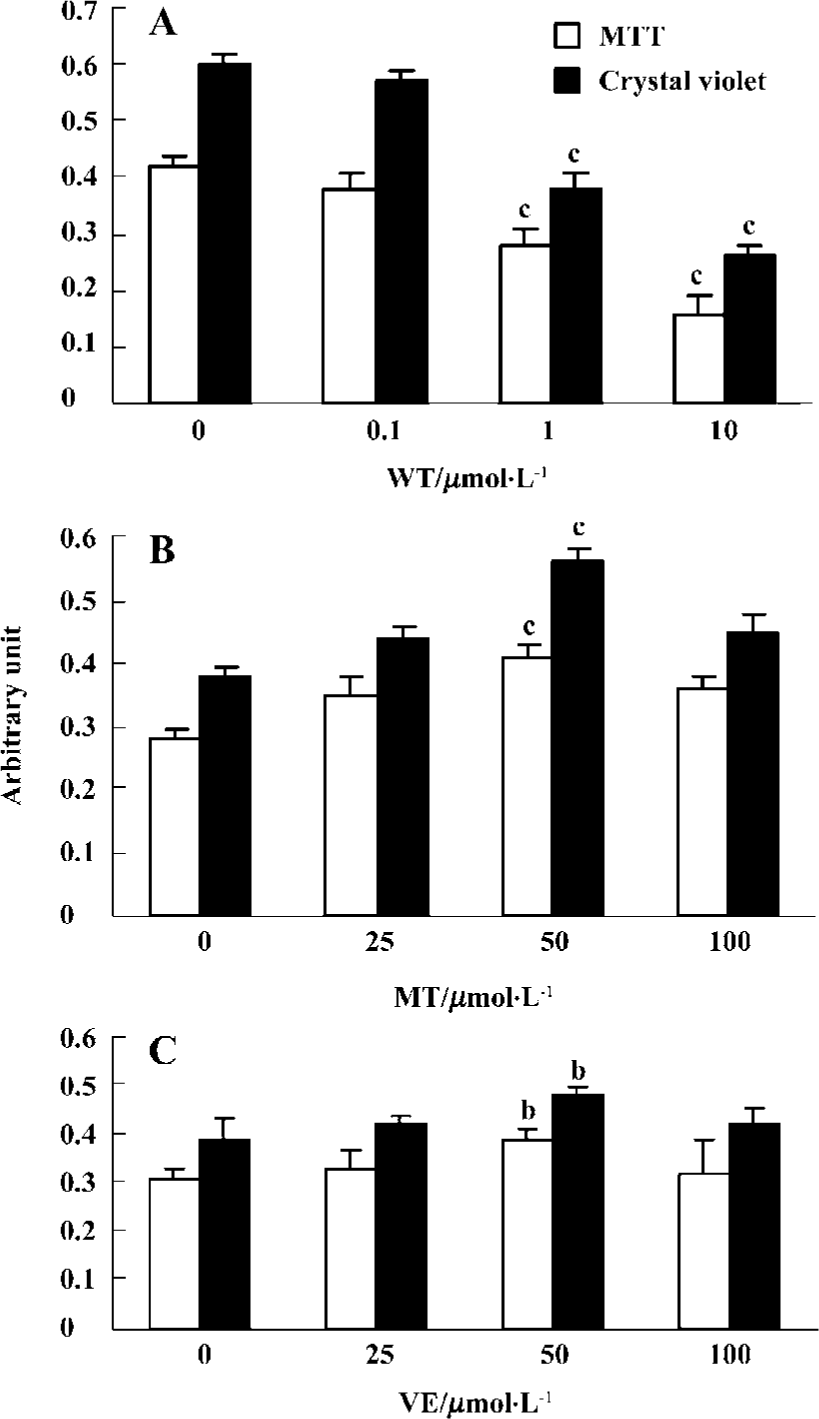
Effect of wortmannin, melatonin, and VE on cell viability and morphology Wortmannin induced decreased cell viability in a concentration-dependent manner from 0.1 µmol/L to 10 µmol/L in 2 h (Figure 1A). Therefore, 1 µmol/L wortmannin, represented 20%-30% inhibition of MTT and crystal violet, was used for all subsequent experiments. Exposure of the cells to 50 µmol/L melatonin (Figure 1B) or 50 µmol/L VE (Figure 1C) before administration of wortmannin resulted in protective effects. However, the premixing of melatonin or VE with wortmannin did not show protection in cell viability (data not shown).
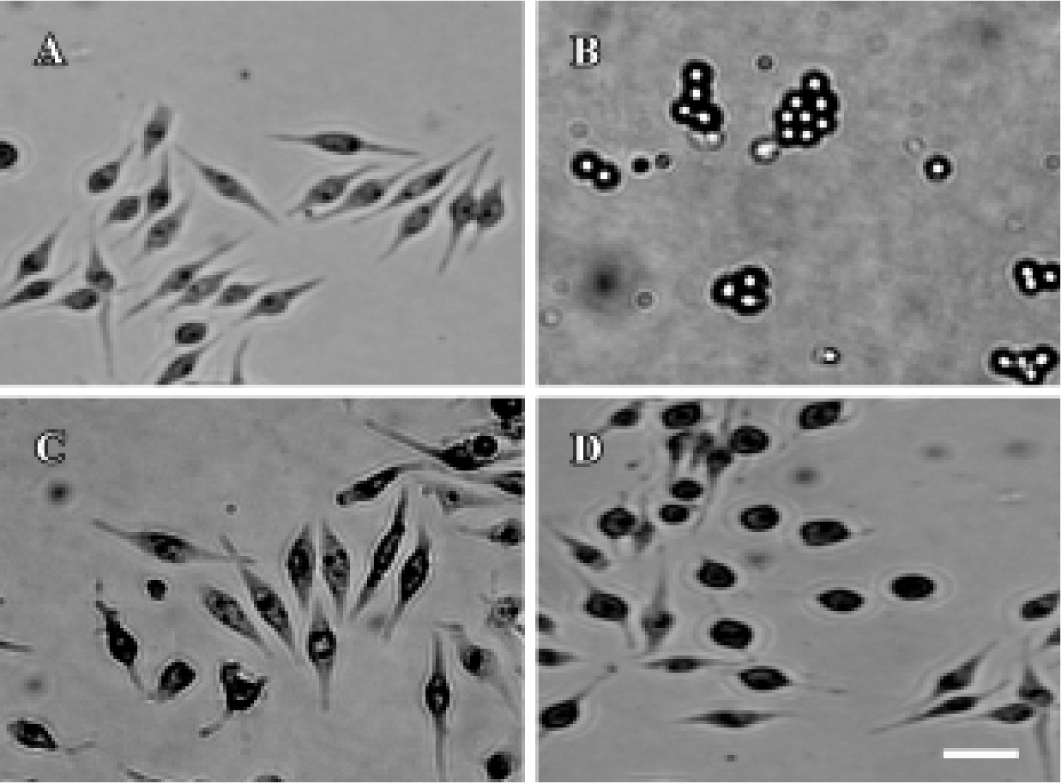
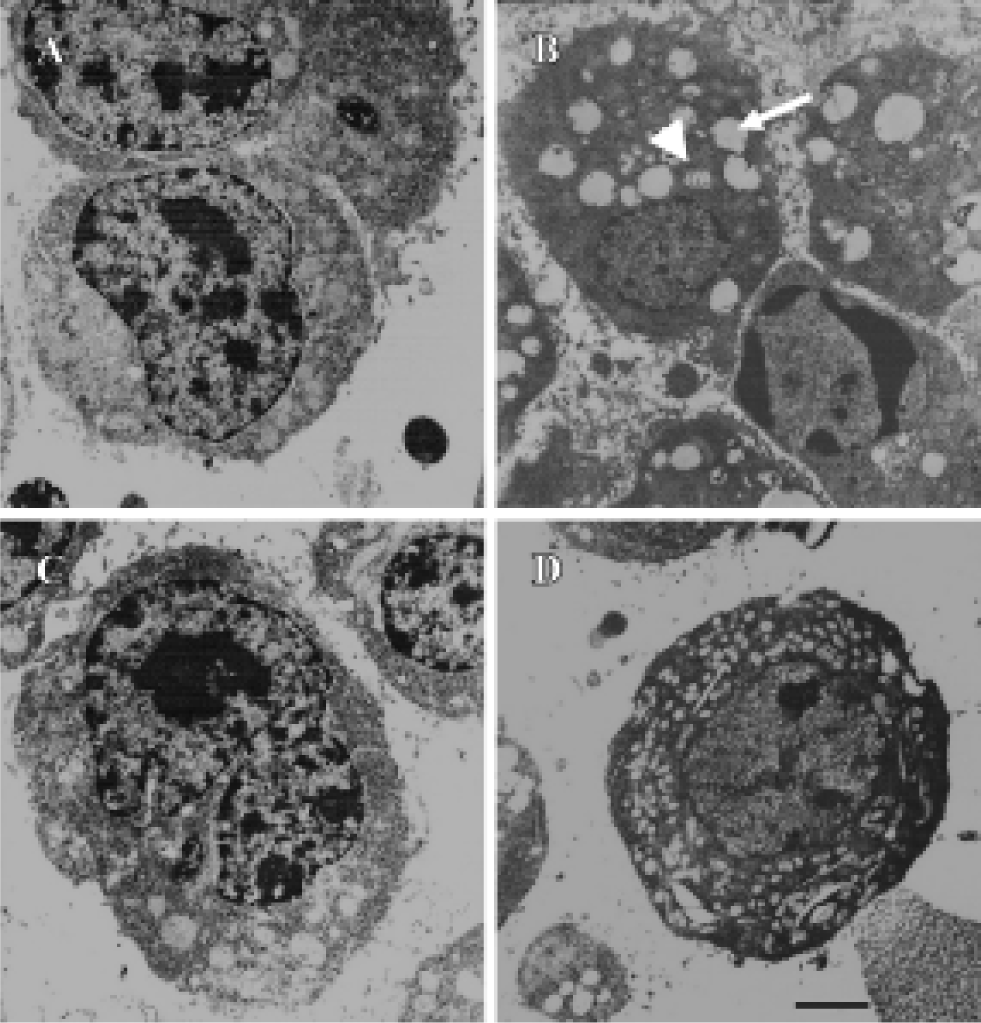
By phase-contrast microscopy, we observed that in the control group the border of the cells was clear with plenty of processes, and treatment of the cells with 1 µmol/L wortmannin induced retraction of cell processes, with some cells detaching from the plate. Preincubation of the cells with 50 µmol/L melatonin or 50 µmol/L VE protected against the morphology changes induced by wortmannin, although the cell body was still larger than normal cells (Figure 2). With electron microscopy, we found that treatment of cells with wortmannin induced organelle vacuolation and mitochondrial swelling (markers of degeneration), but no cellular shrinkage and chromatin condensation (markers of apoptosis) was observed. Pretreatment with melatonin or VE partly protected cells from the degeneration induced by wortmannin (Figure 3).
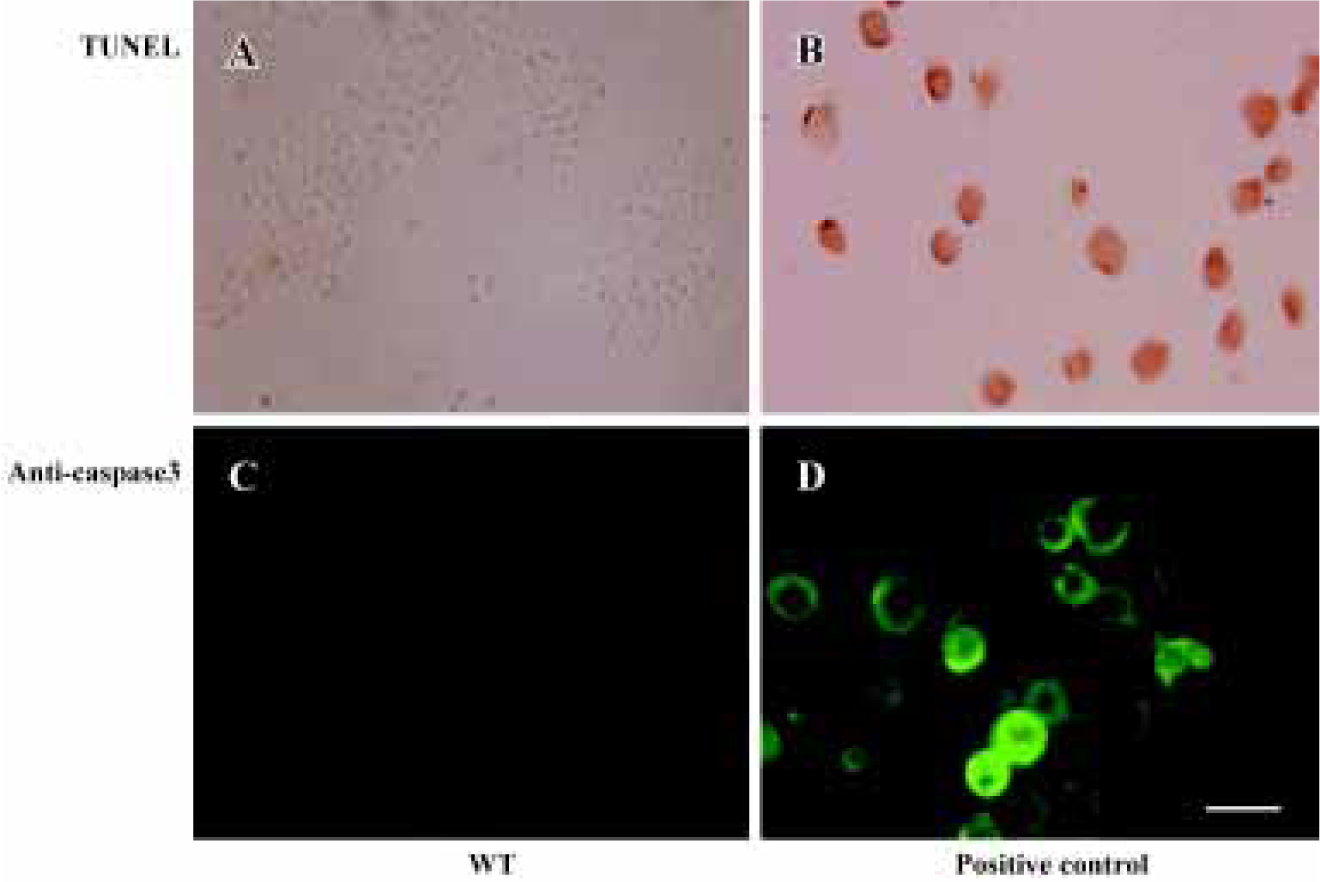
Effect of wortmannin on cell apoptosis Very few positive cells were detected in the 1 µmol/L wortmannin-treated samples. However, numerous apoptotic cells were seen in positive control of 1 µmol/L staurosporine-treated cells (Figure 4).
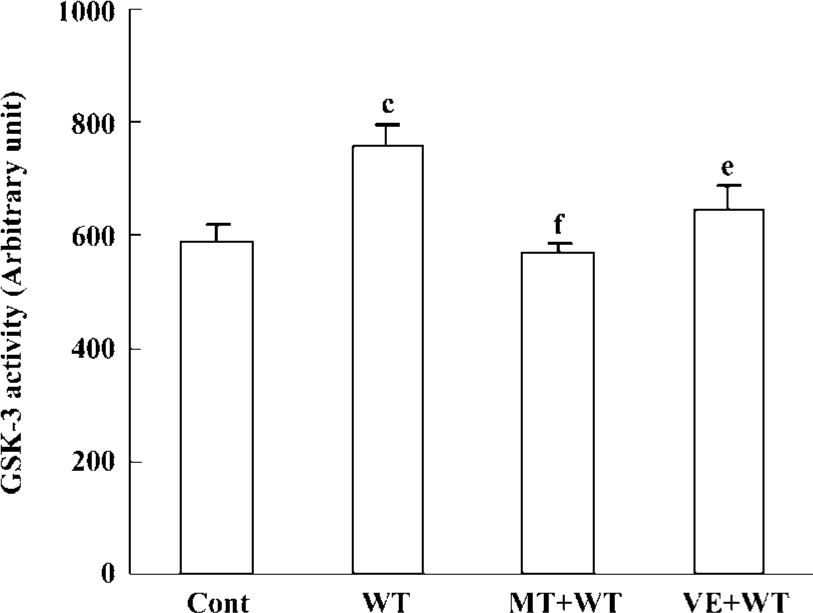
Effect of wortmannin, melatonin, and VE on tau phosphorylation, and the activity of GSK-3β Tau was hyperphosphorylated at both tau-1 (recognized non-phosphorylated tau at Ser-199/202) and PHF-1 (recognized phosphorylated tau at Ser-396/404) epitopes by treatment with wortmannin for 2 h. The hyperphosphorylation of tau was blocked by preincubation of the cells with melatonin and VE. Melatonin showed a more efficient protective effect than VE (Figure 5). We also found that wortmannin increased activity of GSK-3, pretreatment with melatonin almost completely reversed wortmannin-induced activation of GSK-3, and preincubation of VE partially restored the activity of GSK-3 (Figure 6).
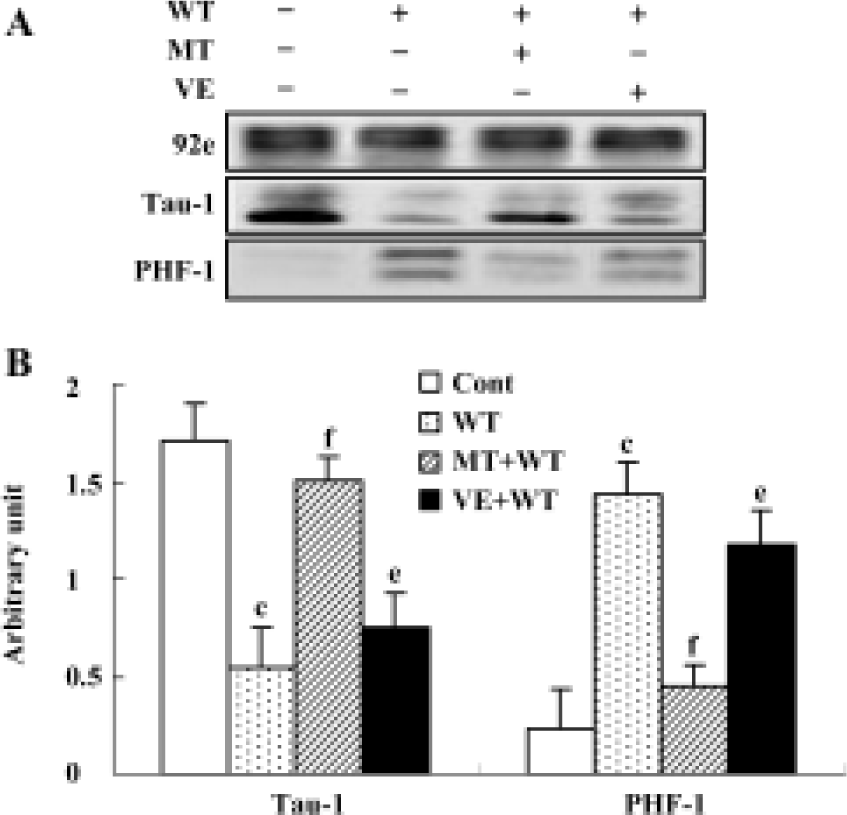
As GSK-3β activity is regulated by the phosphorylation and dephosphorylation on its serine-9 (Ser-9)[16], we further detected the expression of total and phosphorylated GSK-3β. We found that wortmannin treatment did not change the expression of the total GSK-3β, but markedly decreased the expression of phosphorylated-GSK-3 at Ser-9. We also demonstrated that preincubation of melatonin or VE obviously arrested wortmannin-induced decrease of phosphorylated GSK-3β at Ser-9 (Figure7A, 7B).

Effect of wortmannin, melatonin, and VE on lipid peroxidation The level of MDA was significantly higher, but the activity of SOD was significantly lower in wortmannin-treated cells than the vehicle control samples. After preincubation of 50 μmol/L melatonin or VE, the MDA level and SOD activity was partially restored, and the protective effect of melatonin was stronger than that of VE at the same concentration (Table 1).
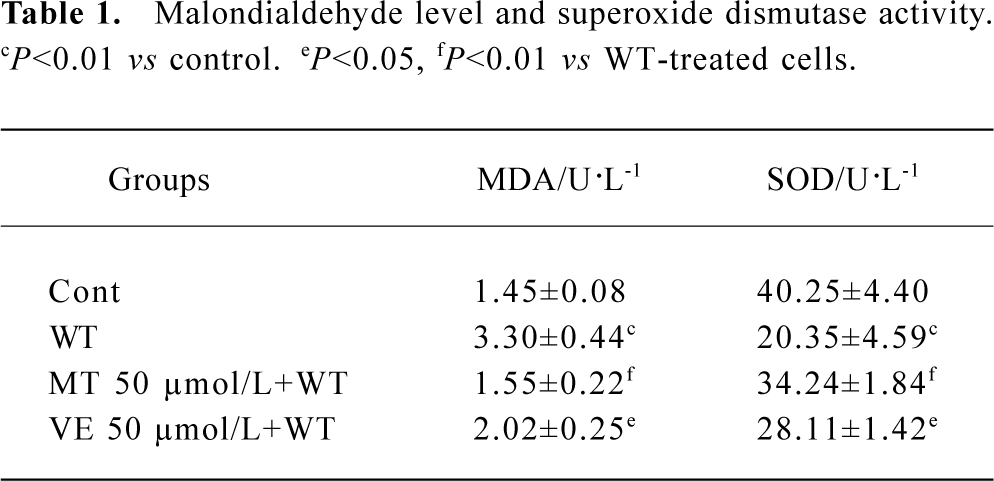
Full table
Discussion
Abnormal hyperphosphorylation of tau is a recognized pathological process, which might be responsible for the disruption of microtubules and thus neurodegeneration seen in the brain of patients with Alzheimer's disease (AD). Many protein kinases are reported to phosphorylate tau at some of the AD sites and can lead to the dysfunction of tau in maintaining stability of cytoskeleton. Among them, GSK-3, a major tau kinase, is a leading candidate for initiating pathologic tau hyperphosphorylation[5,7]. GSK-3 forms a complex with tau in the microtubule fraction from the bovine brain and it is co-localized with phosphorylated tau during development[17]. Hyperphosphorylation of tau by GSK-3 accelerates neurodegeneration and induces fibrillary tau inclusions both in vivo and in vitro[18]. As direct GSK-3 activator is not commercially available at the moment, wortmannin, an inhibitor of phosphatidylinositol 3-kinase (PI3K), has been widely used in activating GSK-3 indirectly through protein kinase B (PKB)-mediated signal transduction pathway; that is, wortmannin turns down PKB through inhibiting PI3K and thus activates GSK-3 by decreasing the phosphorylation level of GSK-3 at its serine residues[6,18,19]. We previously reported that wortmannin could induce Alzheimer's-like hyperphos-phorylation of cytoskeleton proteins in a rat brain[9,20]. To further explore the mechanism of how wortman-nin influences tau phosphorylation, and thus to establish an AD-like cell model, we used wortmannin to treat N2a, a neuroblastoma cell line widely used in the AD field[21-23], in the present study. We have found that wortmannin activates GSK-3 and leads to tau hyperphosphorylation in N2a cell line, demonstrating that wortmannin can be used for reproducing a cell model with AD-like tau hyperphosphorylation. We have also observed that wortmannin treatment induces oxidative stress as displayed by the increase of MDA level and the decrease of SOD activity. This suggests the crucial role of oxidative stress in wortmannin-induced tau hyperphos-phorylation.
Although the treatment of the cells with wortmannin led to significantly decreased cell viability, we did not see a typical cell apoptosis in the present study. The cells show evident hyperphosphorylation of tau, which is considered to be one of the earliest signs of neuronal degeneration and appears to precede tau aggregation or amyloid formation[24]. Tau abnormality provides the basis for the unequivocal diagnosis. However, its role in AD pathogenesis, especially in cell loss, is still not understood. The results observed in this study are consistent with the observations of SY5Y cells that tau phosphorylation can be an anti-apoptosis[25]. In another project, we also found that treatment of cells with calyculin A (a protein phosphatase inhibitor) led to neurofila-ment hyperphosphorylation and aggregation with minimal apoptosis[12]. Neurons in AD were not co-labeled with AT8 (an antibody that probes phosphorylated tau) and an antibody to activate caspase-3[26]. Similarly, activated JNK, p38, and ERK1/2 (phosphorylated forms of these proteins) co-localized with hyperphosphorylated tau in an AD brain, but not with markers of apoptosis[27,28], indicates that increased tau phosphorylation seems to resist apoptotic stimuli. Therefore, although many in vivo and in vitro data support the involvement of apoptosis in neurodegenerative pro-cesses, there is considerable evidence suggesting that very complex events can contribute to neuronal death with possible repair mechanisms[29], elucidation of which could prove useful for future investigation of the mechanisms of cell death in these disorders and their relations to cytoskeletal abnormalities, as well as prevention and therapy of neuro-degenerative disorders.
AD is the most common cause of dementia and there is no specific and effective cure for this disorder to date. Recently, increasing data suggests that melatonin might play an important role in the development of AD and serve as a candidate for arresting AD-like pathological processes[30,31]. Patients with AD have a more profound reduction of melatonin in the brain and in cerebrospinal fluid[31,32]. The efficacy of melatonin in inhibiting oxidative damage has been tested in a variety of neurodegenerative disease models, including Huntington's disease, Parkinson's disease and AD, induced with quinolinic acid, MPTP, β-amyloid, and OA (okadaic-acid) respectively in rat brain or in cultured cells[33-36]. In the present study, we have found that melatonin rescues cell viability and reverses tau hyperphosphorylation induced by wortman-nin in N2a neuroblastoma cells. At the same time, melatonin decreases the level of lipid peroxide and increases the activity of antioxidant enzymes. This is consistent with previous findings that melatonin prevents oxidative damage to the cell membrane, organelles, nuclear and mitochondrial DNA by donating electrons[37]. From this data, we speculate that melatonin might prevent wortmannin-induced tau hyper-phosphorylation through its antioxidant effects. We have also found that melatonin obviously inhibits wortmannin-induced activation of GSK-3, suggesting that melatonin might act not only against free radicals, but also indirectly as an enzyme modulator[31] in rescuing wortmannin-induced tau hyperphosphorylation; further study is needed to explore the mechanism. We also observed that melatonin acted more efficiently than vitamin E not only as an antioxidant but also in restoring GSK-3 activity and tau hyperphosphorylation. Melatonin is a small amino acid hormone secreted mainly in the pineal gland. Specific characters of melatonin, such as its solubility in both lipids and water that allow it to be easily penetrated into the cells and pass through the blood brain barrier, can make it totally different from other antioxidant. In addition to its role in rescuing AD-like tau abnormalities, melatonin is also reported to protect β-amyloid peptide-induced cell injuries[38,39]. Therefore, melatonin might become a promising candidate thwarting the two hallmark abnormalities in AD patients.
Melatonin could function through its membranous and nuclear receptors in peripheral tissues; melatonin receptors are widely expressed in different organs, such as the brain hippocampus, retina, lung, liver, and kidney[40]. Melatonin could also function through non-receptor mediated pathways[41]. In N2a cells, whether melatonin functions through the receptor-dependent pathway or receptor-independent pathway, needs further study.
Taken together, we have found in the present study that wortmannin induces overactivation of GSK-3 and tau hyperphosphorylation with a concurrent lipid peroxidation and reduction of cell viability in the N2a cell line. Melatonin rescues not only wortmannin-induced oxidative stress, but also GSK-3 overactivation and tau hyperphosphorylation.
References
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001;24:1121-59.
- Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimers Dis 1999;1:329-51.
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interac-tion, intracellular trafficking and neurodegeneration. Biochem J 1997;323:577-91.
- Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 2000;275:15733-40.
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 2004;89:1313-7.
- Li X, Bijur GN, Jope RS. Glycogen synthase kinase-3β, mood stabilizers, and neuroprotection. Bipolar Disord 2002;4:137-44.
- Bhat RV, Budd SL. GSK-3beta signalling: casting a wide net in Alzheimer抯 disease. Neurosignals 2002;11:251-61.
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, et al. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol 1999;58:1010-9.
- Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, et al. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphos-phorylation of tau and impairment of spatial memory. J Neurochem 2003;87:1333-44.
- Reiter RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev 1991;12:151-80.
- Zhang YC, Wang ZF, Wang Q, Wang YP, Wang JZ. Melatonin attenuates beta-amyloid-induced inhibition of neurofilament expression. Acta Pharmacol Sin 2004;25:447-51.
- Li SP, Deng YQ, Wang XC, Wang YP, Wang JZ. Melatonin protects SH-SY5Y neuroblastoma cells from calyculin A-induced neurofilament impairment and neurotoxicity. J Pineal Res 2004;36:186-91.
- Wang DL, Ling ZQ, Cao FY, Zhu LQ, Wang JZ. Melatonin attenuates isoproterenol-induced protein kinase an overactivation and tau hyperphosphorylation in rat brain. J Pineal Res 2004;37:11-6.
- Kim BC, Shon BS, Ryoo YW, Kim SP, Lee KS. Melatonin reduces X-ray irradiation-induced oxidative damages in cultured human skin fibroblasts. J Dermatol Sci 2001;26:194-200.
- Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 1974;47:469-74.
- Lee CW, Lau KF, Miller CC, Shaw PC. Glycogen synthase kinase-3 beta-mediated tau phosphorylation in cultured cell lines. Neuroreport 2003;14:257-60.
- Sun W, Qureshi HY, Cafferty PW, Sobue K, Agarwal-Mawal A, Neufield KD, et al. Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J Biol Chem 2002;277:11933-40.
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med 2002;8:126-32.
- Krasilnikov MA. Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation. Biochemistry (Mosc) 2000;65:59-67.
- Liu SJ, Wang JZ. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol Sin 2002;23:183-7.
- Wang YP, Wang ZF, Zhang YC, Qing Tian, Wang JZ. Effect of amyloid peptides on serum withdrawal-induced cell differentiation and cell viability. Cell Res 2004;14:467-72.
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 2002;156:1051-63.
- Munoz JP, Alvarez A, Maccioni RB. Increase in the expression of the neuronal cyclin-dependent protein kinase cdk-5 during differentiation of N2A neuroblastoma cells. Neuroreport 2000;11:2733-8.
- Brandt R. Cytoskeletal mechanisms of neuronal degeneration. Cell Tissue Res 2001;305:255-65.
- Lesort M, Blanchard C, Yardin C, Esclaire F, Hugon J. Cultured neurons expressing phosphorylated tau are more resistant to apoptosis induced by NMDA or serum deprivation. Brain Res Mol Brain Res 1997;45:127-32.
- Jellinger KA. Cell death mechanisms in neurodegeneration. J Cell Mol Med 2001;5:1-17.
- Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, et al. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol 2001;60:1190-7.
- Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 2001;11:144-58.
- Marx J. Neuroscience. New leads on the‘how’of Alzheimer's disease. Science 2001;293:2192-4.
- Grundman M, Grundman M, Delaney P. Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc 2002;61:191-202.
- Matsubara E, Shoji M, Murakami T, Kawarabayashi T, Abe K. Alzheimer's disease and melatonin. Int Congress Series 2003;1252:395-98.
- Beyer CE, Steketee JD, Saphier D. Antioxidant properties of melatonin朼n emerging mystery. Biochem Pharmacol 1999;57:1077.
- Guillemin GJ, Brew BJ. Implications of the kynurenine pathway and quinolinic acid in Alzheimer's disease. Redox Rep 2002;7:199-206.
- Cabrera J, Reiter RJ, Tan DX, Qi W, Sainz RM, Mayo JC, et al. Melatonin reduces oxidative neurotoxicity due to quinolinic acid: in vitro and in vivo findings. Neuropharmacology 2000;39:507-14.
- Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, et al. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer's disease. Prog Neurobiol 2000;62:633-48.
- Montilla-Lopez P, Munoz-Agueda MC, Feijoo Lopez M, Munoz-Castaneda JR, Bujalance-Arenas I, Tunez-Finana I. Comparison of melatonin versus vitamin C on oxidative stress and antioxidant enzyme activity in Alzheimer's disease induced by okadaic acid in neuroblastoma cells. Eur J Pharmacol 2002;451:237-43.
- Reiter RJ. Melatonin. Lowering the high price of free radicals. News Physiol Sci 2000;15:246-50.
- Matsubara E, Bryant-Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, Herbert D, et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer's disease. J Neurochem 2003;85:1101-8.
- Reiter RJ, Tan DX, Sainz RM, Mayo JC, Lopez-Burillo S. Melatonin: reducing the toxicity and increasing the efficacy of drugs. J Pharm Pharmacol 2002;54:12299-321.
- Naji L, Carrillo-Vico A, Guerrero JM, Calvo JR. Expression of membrane and nuclear melatonin receptors in mouse peripheral organs. Life Sci 2004;74:2227-36.
- Pappolla MA, Simovich MJ, Bryant-Thomas T, Chyan YJ, Poeggeler B, Dubocovich M, et al. The neuroprotective activities of melatonin against the Alzheimer beta-protein are not mediated by melatonin membrane receptors. J Pineal Res 2002;32:135-42.