
Pharmacokinetic behaviors and oral bioavailability of oridonin in rat plasma1
Introduction
Rabdosia rubescences (Chinese name “Donglingcao”), a herbal medicine, is traditionally used in China for the treatment of tonsillitis and a variety of cancers. Oridonin, a diterpenoid extracted from Rabdosia rubescences, is the marker compound and one of the major antitumor components of this herb[1,2]. Oridonin injection is used alone or in combination with other drugs to treat human cancers, especially for the treatment of liver cancer[3,4], esophageal carcinoma and carcinoma of gastric cardia[5]. Oridonin injection and Rabdosia rubescences can also extend the lives of advanced cancer patients as well as improve their living quality. Due to the low toxicity of oridonin and Rabdosia rube- scences, they can be used at a high dose for a long period of time, and only a few patients suffer abdominal discomfort[5]. Because of the abundance of Rabdosia rubescences in China, recently, oridonin and Rabdosia rubescences have attracted special attention.
Although Rabdosia rubescences and oridonin have been used clinically for a long time, there is little information in published literature on the pharmacokinetics of oridonin. To our knowledge, only the pharmacokinetics of oridonin after iv administration in rabbits and iv and ip administration in mice have been reported[6,7], and there is no published information about the pharmacokinetics of oridonin following oral administration. In clinical trials, Rabdosia rubescences is usually administered orally, so a better understanding of the pharmacokinetics and oral bioavailability of oridonin is very important for explaining the therapeutical outcomes produced by the drug in clinical trials and to help establish a rational dosage regimen.
In the present paper, we describe the pharmacokinetics behavior of oridonin after intravenous, oral and intraperitoneal administrations to rats.
Materials and methods
Chemicals and reagents Oridonin (98.9%) was extracted from the aerial parts of Rabdosia rubescences and refined in our laboratory (identified by 1H-NMR, UV and MS). The internal standard, ethyl hydroxybenzoate (99.5%), was supplied by Shenyang Dongxing Reagent Factory (Shenyang, China). HPLC-grade methanol was obtained from Concord Tech Co (Tianjin, China) while HPLC-grade ethyl acetate and n-butanol were from purchased from Tianjin Kermel Chemical Reagents Development Centre (Tianjin, China). All other reagents were of analytical grade. Distilled water, prepared from deionized water, was used throughout the study.
Oridonin solution For intravenous, oral and intraperitoneal administration, oridonin (5 mg/mL) solution was prepared in 0.9% (w/v) saline containing 30% ethanol (v/v).
Animals and surgical procedures Male Wistar rats (230–250 g) were supplied by the Lab Animal Center of Shenyang Pharmaceutical University (Shenyang, China). All experimental procedures were carried out in accordance with the guidelines of the Experimental Animal Care and Use Committee of Shenyang Pharmaceutical University. The animals were maintained under standard laboratory conditions on a 12 h light/dark cycle and were fed standard rat chow and water ad libitum. The rats fasted overnight before the experiments and food was returned 2 h after dosing. Water was available ad libitum throughout the experiments.
Drug administration and sample collection Three groups of rats (each group contained 6 rats) were given oridonin solution as a single dose of 5, 10 and 15 mg/kg via the femoral vein (slightly anaesthetized by aether). The infusion time was about 10 s. Blood samples (250 µL) were collected in heparinized tubes from orbit veins with an heparinized glass tube at 0.083, 0.167, 0.5, 1, 2, 4, 6, 9, 12, 24, 36, and 48 h after administration; another 3 groups of rats (each group contained 6 rats) received a single dose of 20, 40, and 80 mg/kg by oral gavage. Blood samples (250 µL) were collected in the heparinized tubes from each rat at 0.05, 0.1, 0.167, 0.25, 0.5, 1, 2, 4, 6, 9, 12, 24, 36, and 48 h after administration. Six rats were given oridonin solution as single dose of 10 mg/kg intraperitoneally and blood samples (250 µL) were collected in the heparinized tubes from each rat at 0.083, 0.167, 0.5, 1, 2, 4, 6, 9, 12, 24, and 36 h after administration. Blood samples were immediately centrifuged and stored at -20 °C until analysis.
Sample preparation and analysis The concentrations of oridonin in rat plasma were determined by an HPLC/electrospray ionization mass spectrometric detection (HPLC/ESI-MS) method developed and validated in our laboratory[8]. Briefly, to 100 µL plasma in glass centrifuge tubes 50 µL ethyl hydroxybenzoate (internal standard, 80 ng/mL) and 50 µL mobile phase were added. Samples were then vortex-mixed for 30 s and extracted with 3 mL ethyl acetate-n-butyl alcohol (100:2, v/v). After vortex-mixing for 1 min and shaking for 10 min, the organic and aqueous phases were separated by centrifugation at 2000×g for 10 min, then the upper organic layer was transferred to another tube and evaporated to dryness at 40 °C under a gentle stream of nitrogen. The residue was reconstituted in 100 µL mobile phase followed by vortex-mixing and centrifugation at 2 000×g for 10 min. Then, 20 µL of an aliquot of the supernatant was injected onto the HPLC/ESI-MS system.
The high-performance liquid chromatography was performed using a Waters 1525 Binary pump (Framingham, Massachusetts, USA), which was controlled by Masslynx 4.0 Software (Waters Corp, Framingham, Massachusetts, USA). The mobile phase consisted of methanol-water (80:20, v/v) at a flow rate of 1.0 mL/min and the injection volume was 20 µL. The analytical column used was a DiamonsilTM C18 column (200 mm×4.6 mm id, 5 µm) from Dikma Tech (Beijing, China) at a column temperature of 25 °C.
A ZQ2000 micromass spectrometer (Waters Corp, USA) fitted with a Z-Spray ion interface was used for all analyses. Ionization was achieved by using electrospray in the negative mode. The following parameters were optimized for the analysis of oridonin: capillary voltage, 3.0 kV; cone voltage, 25 V; source temperature, 105 °C; and desolvation gas (nitrogen) heated to 350 °C and delivered at a flow rate of 350 L/h. Quantification was performed using the selected ion recording of m/z 363 for oridonin and m/z 165 for ethyl hydroxybenzoate. The lower limit of quantification of the method was 10 ng/mL and the quantitation range was 10–4 000 ng/mL. For samples containing oridonin at a concentration higher than the upper limit of the range in the standard curve, an aliquot of the sample was first diluted with blank rat plasma and then 100 µL of the diluted sample was treated as described. The intraday and interday accuracy and precision of the assay were less than 9%.
Pharmacokinetic analysis All pharmacokinetic parameters were determined by non-compartmental analysis. The peak plasma level (Cmax) and the time to reach the peak plasma concentration (tmax) were obtained directly from the concentration-time data. The elimination rate constant (Ke) was calculated from the slope of the logarithm of the plasma concentration versus time using the final 4 points. The apparent elimination half life (t1/2) was calculated as 0.693/Ke. The area under the plasma concentration-time curve (AUC) and the area under the first moment curve (AUMC) were calculated by the trapezoidal rule. Total body clearance was calculated as X0/AUC. The extent of absolute bioavailability was estimated from the dose-normalized ratios of (AUC0-∞)po to (AUC0-∞)iv (based on iv 10 mg/kg). The mean residence times after intravenous administration (MRTiv) and oral administration (MRTpo) were calculated by dividing the AUMC by the AUC. The mean absorption time (MAT) was calculated by subtracting MRTiv from MRTpo. The values were calculated by Microsoft Excel (Microsoft, Seattle, Washington, USA) and each value was expressed as mean±SD.
Results
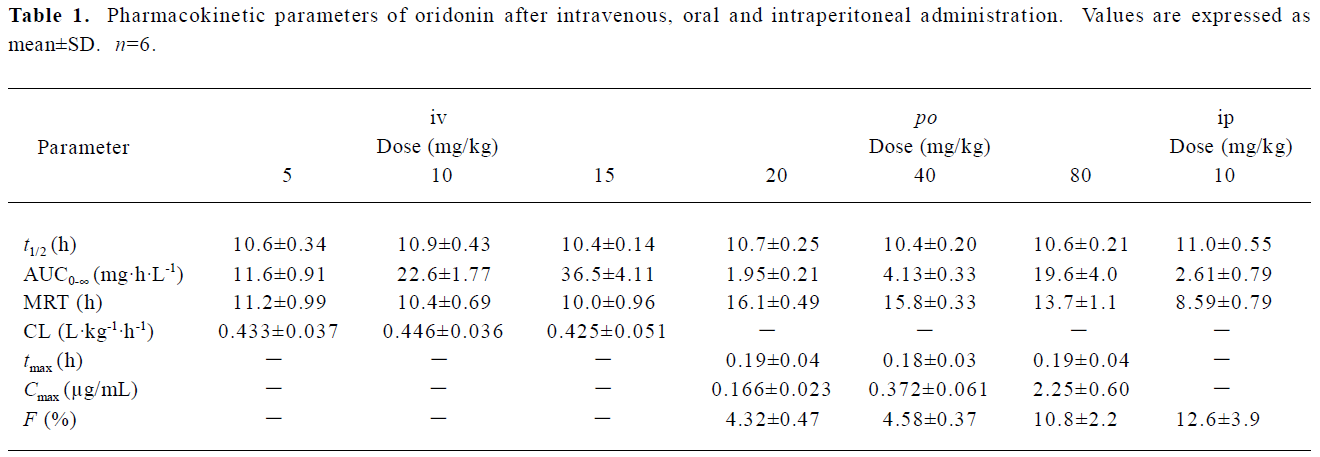
Pharmacokinetic analysis of plasma concentrations after intravenous administration After intravenous administration, the plasma concentration of oridonin first decreased rapidly and then more slowly, that is to say, the plasma concentration of oridonin after intravenous administration decreased polyexponentially and the terminal elimination half life was relatively long (about 10 h). After intravenous administration, the pharmacokinetic parameters of oridonin were dose-independent at 3 doses: 5, 10, and 15 mg/kg. These results show that oridonin exhibits linear kinetics following intravenous administration over the dose range studied (Table 1, Figure 1).
Full table
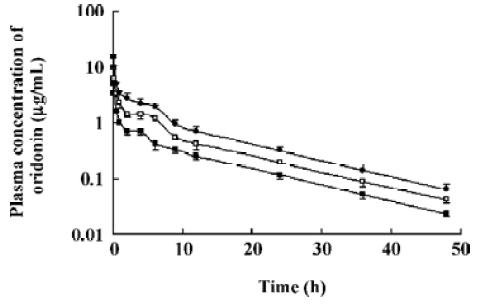
Pharmacokinetic analysis of plasma concentrations after oral administration The mean plasma concentration versus time curve of oridonin increased rapidly after oral administration reaching the maximum level less than 15 min after administration. Starting 9 h after administration, the profiles of oral and intravenous administration declined in parallel. The mean plasma concentration versus time curves after intravenous administration was much higher than that after oral administration at a higher dose. The extent of absolute bioavailability was rather low and appeared to be dose dependent. At a high dose (80 mg/kg), the extent of absolute oral bioavailability was about 2 times higher than those of low doses (20 and 40 mg/kg) (Table 1, Figure 2).
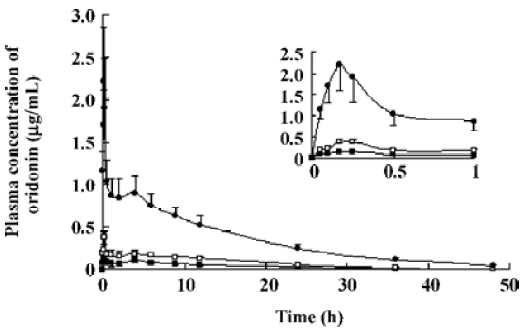
Pharmacokinetic analysis of plasma concentrations after intraperitoneal administration After intraperitoneal administration, the plasma concentration-time curve was similar to that after intravenous administration, except that the plateau was absent. Furthermore, the extent of absolute bioavailability of oridonin was only 12.6% (Figure 3).
Discussion
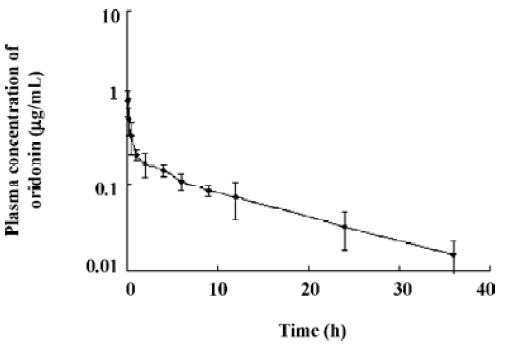
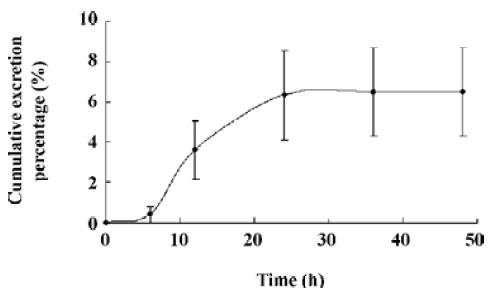
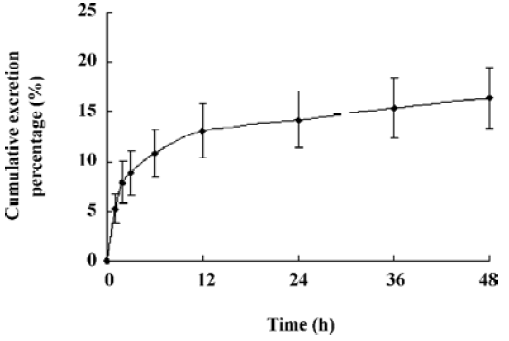
The oral absolute bioavailability of oridonin was rather low (4.32%–0.8%) and appeared to be dose dependent. Considering the amount of unchanged oridonin recovered from the gastrointestinal tract and feces 48 h after oral administration (the mean value was approximately 6.52%; Figure 4), the low extent of absolute oral bioavailability values are most likely due to hepatic, gastric and/or intestinal first-pass effects. After intraperitoneal administration of oridonin solution to rats at a single dose of 10 mg/kg, the bioavailability of oridonin was 12.6%; Figure5). This result shows that hepatic first-pass effect may be the main reason for the low oral bioavail-ability of oridonin. Oridonin is a water-insoluble drug; some researchers have tried to enhance its oral bioavailability by simply increasing the dissolution rate of the drug from its dosage form[9]. Although in vivo behavior of those dosage forms had not been performed, based on the result of our paper we can speculate that through those meanings, the enhanced oral bioavailability is limited. Other drug delivery systems which can circumvent the liver first-pass effect may work, such as the M cell drug delivery system[10] (Table 1, Figure 3).
Many natural products display beneficial anticancer effects in vitro, but only a few have been involved in clinical trials. One of the main obstacles which prevent the development of natural anticancer drugs is that the concentration of drugs in vivo can not reach the level used in vitro. The in vitro anticancer effect of oridonin has been studied by several groups[11–16]. These research show that the anticancer effect of oridonin is time and dose-dependent; at a low µmol/L concentration (about 1 µg/mL), oridonin has weak apoptosis-inducing effects, while at a concentration of 8–10 µmol/L, oridonin has strong apoptosis-inducing effects on most cancer cells[11,13]. According to the clinical dose of Rabdosia rubescences[17] and the content of oridonin in the herb[1], a patient receiving about 175 mg oridonin per time orally is equal to the dose of 13.5 mg/kg to rats[18]. Obviously at this dose the plasma concentration of oridonin is too low to have a beneficial effect. In clinical trials, patients receive Rabdosia rubescences solution 3 times per day. If the similar poor and dose-dependent oral bioavailability phenomenon is found in men, a higher dose and a longer dosing interval may produce better curative effects.
In clinical trials, Rabdosia rubescences and oridonin are usually used in combination with other drugs to enhance the anticancer effects of chemical drugs[17,19]. Since the hepatic first-pass effect may be the main reason for the low oral bioavailability of oridonin, potential drug-drug interactions may occur: enhanced oral bioavailability of oridonin and decreased drug metabolism, which may lead to better anticancer effects or more severe side effects.
The pharmacokinetic parameters of oridonin were dose independent at 3 doses, 5, 10, and 15 mg/kg after intravenous administration. The dosage range was set according to the toxicity of the menstruum and the sensitivity of our analytical method, but the dosage range is somewhat narrow to give more pharmacokinetic information. Considering that the length of the plateaus of the 3 curves in Figure 1 look dose dependent, we wonder what will happen with a reduction of the dose. Although an intravenous administration experiment at an even lower dose was not carried out, the intraperitoneal administration experiment may be used as a reference. After intraperitoneal administration of oridonin solution to rats at a single dose of 10 mg/kg, the plateau of plasma concentration-time disappeared. Since the percentages of the intravenous dose of oridonin excreted in bile as unchanged drug was 16.0% in the rats (data not shown), the enterohepatic circulation may contribute to the plateau in the plasma concentration-time curves of Figure 1 and Figure 2. The plateaus of the curves in Figure 1 may also be accounted by saturable tissue uptake since the sharp decrease of the plasma oridonin concentration after intravenous administration was due to tissue uptake.
In conclusion, we systematically investigated the pharmacokinetic behaviors of oridonin after intravenous, oral and intraperitoneal administration in rats. The mean plasma concentration-time curve appeared to be polyexponential, and oridonin exhibited linear kinetics at 3 doses following intravenous administration. The extent of absolute oral bioavailability was rather low and dose-dependent.
Acknowledgments
The authors would like to thank Dr Feng QIU and Dr Liang CUI for the technical help in animal experiments.
References
- Yuan K, Yang ZH, Yang Y, Zhang XM, Sun W. Determination of oridonin in Rabdosia rubescences by HPLC. Henan Sci 1998;16:175-7. Chinese..
- Zuo HJ, Li D, Wu B, Gao HY, Wu LJ, Ikejima T. Studies on the constituents of Rabdosia rubescens (Hemsl) Hara and their antitumor activities in vitro. J Shenyang Pharm Univ 2005;22:258-62. Chinese..
- Guan YZ, Wei TH. Clinical research on the oridonin injection for the interventional therapy of liver cancer. J Med Radiol Technol 2005;236:43. Chinese..
- Wang RL. Clinical evaluation of Rabdosia rubescences for 31 patients with liver cancer. Chin J Cancer 1984;8:50. Chinese..
- Henan Research Group for. Rabdosia rubescences. Conclusion of clinical evaluation of Rabdosia rubescences for the treatment of 68 patients with esophageal carcinoma and carcinoma of gastric cardia. Cancer Res Prev Treat 1976;3:32-3. Chinese..
- Zhang DR, Ren TC, Lou HX, Xing J. The tissue distribution in mice and pharmacokinetics in rabbits of oridonin-solid lipid nanoparticles. Acta Pharm Sin 2005;40:573-6. Chinese..
- Lin C, Zhang TM, Wu WK, Cao SH, Wang DX, Yi MG. Absorption, distribution and excretion of [3H]rubescensine A in mice. Acta Pharmacol Sin 1983;4:57-60. Chinese..
- Xu W, Sun J, Zhang TT, Tang JL, Li HY, Fang JL, et al. A rapid and accurate HPLC/ESI-MS method for the quantitative determination of oridonin in rat plasma. Pharmazie 2006;61:757-9.
- Zhang YB, Kou X, Lu JS, Wang GH. Study on the inclusion of oridonin-β-cyclodextrin. J Chin Med Mater 1999;22:204-5. Chinese..
- Jepson MA, Clark MA, Hirst BH. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv Drug Deliv Rev 2004;56:511-25.
- Liu JJ, Wu XY, Peng J, Pan XL, Lu HL. Antiproliferation effects of oridonin on HL-60 cells. Ann Hematol 2004;83:691-5.
- Heish TC, Wijeratne EK, Liang JY, Gunatilaka AL, Wu JM. Differential control of growth, cell cycle progression, and expression of NF-κB in human breast cancer cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin, diterpenoids from the Chinese herb Rabdosia rubescens. Biochem Biophys Res Commun 2005;337:224-31.
- Ren KK, Wang HZ, Xie LP, Chen DW, Liu X, Sun J, et al. The effects of oridonin on cell growth, cell cycle, cell migration and differentiation in melanoma cells. J Ethnopharmacol 2006;103:176-80.
- Chen S, Gao J, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. The cytostatic and cytotoxic effects of oridonin (Rubescenin), a diterpenoid from Rabdosia rubescens, on tumor cells of different lineage. Int J Oncol 2005;26:579-88.
- Ikezoe T, Yang Y, Bandobashi K, Saito T, Takemoto S, Machida H, et al. Oridonin, a diterpenoid purified from Rabdosia rubescens, inhibits the proliferation of cells from lymphoid malignancies in association with blockade of the NF-kappa B signal pathways. Mol Cancer Ther 2005;4:578-86.
- Huang J, Wu L, Tashiro S, Onodera S, Ikejima T. A comparison of the signal pathways between the TNF alpha- and oridonin-induced murine L929 fibrosarcoma cell death. Acta Med Okayama 2005;59:261-70.
- Wang RL, Fan KS, Fan QX, Ji ZC, Yue BY, Chen ST, et al. Clinical evaluation for 437 esophageal carcinoma patients. Chin J Integrat Tradit West Med 1989;9:740-1. Chinese..
- Xu SY, Bian RL, Chen X. Methodology for pharmacological experiments, 3rd ed. Beijing: People’s Medical Publishing House; 2001.
- Cai RC, Guo HF. Clinical research of Donglingcao combined with UFT for the treatment of advanced lung cancer. Prac Clin J Integr Tradit Chin Western Med 2003;3:12-3. Chinese..