
Antisense oligonucleotides targeting midkine induced apoptosis and increased chemosensitivity in hepatocellular carcinoma cells
Introduction
Midkine (MK) was first identified in embryonal carcinoma cells in the early stages during retinoic acid-induced differentiation[1]. MK and pleiotrophin (also known as the heparin-binding growth-associated molecule) comprise a family of heparin-binding growth/differentiation factors, different from other heparin-binding growth factors such as fibroblast growth factor and hepatocyte growth factor[2–4]. It has been reported that MK is overexpressed in various malignant tumors, such as lung, breast, esophageal, gastric, colorectal, liver, pancreatic, ovarian, urinary bladder, prostatic, cerebral and renal malignancies[5–13], whereas in normal adult tissues, MK is low or undetectable[14]. High expression of MK is correlated with poor prognosis for patients with urinary bladder carcinomas, glioblastomas and neuroblastomas[9,11,12]. Elevated blood MK has also been reported for esophageal cancer, neuroblastoma and gastric cancer[15–17]. MK promotes the survival[18,19], growth[20,21] and migration[22-24] of many cells, which in combination may contribute to oncogenesis and tumor progression. MK can activate mitogen-activated protein kinase (MAPK) pathways through inducing the phosphorylation of p44/42 MAPK (Thr202/Tyr204), subsequently promoting cell growth[25]. MK also activates extracellular signal-regulated kinases 1 and 2, which are well known as signal transducers acting downstream several receptors[18]. The activated MAPK pathway was reported to downregulate caspase-3 activity in neurons[25]. In addition, it was also reported that MK can induce phosphorylation of protein kinase B(AKT) (Ser473 and Thr308). The phosphorylated AKT then promotes a series of anti-apoptosis pathways in cells.
Hepatocellular carcinoma (HCC) is one of the most common malignancies. Several mechanisms of carcinogenesis have been proposed for HCC[26-29]. Downregulation of apoptosis and accelerated proliferative activity in hepatic cells have been regarded as malignant factors leading to HCC[30]. Recently, HCC tumor cells were found to overexpress MK[31]. These findings suggest that MK could be a potential target for hepatocellular carcinoma therapy.
By inducing degradation of the mRNA encoding the protein, oligonucleotides have the potential for cancer therapy[32]. The present study aims to develop potential antisense oligonucleotides (ASODN) to repress MK expression in hepatocellular carcinoma cells and examine their antiproliferation activity.
Materials and methods
Antisense oligodeoxynucleotides All the antisense compounds used in these experiments were designed by a computational neural network model[33,34]. BLAST confirmed that they are specific for MK mRNA. 20 mer ASODN were synthesized using an applied Biosystems 391 DNA synthesizer and purified by OPC (Perkin-Elmer, Foster City, CA, USA). Table 1 shows the sequence of ASODN.
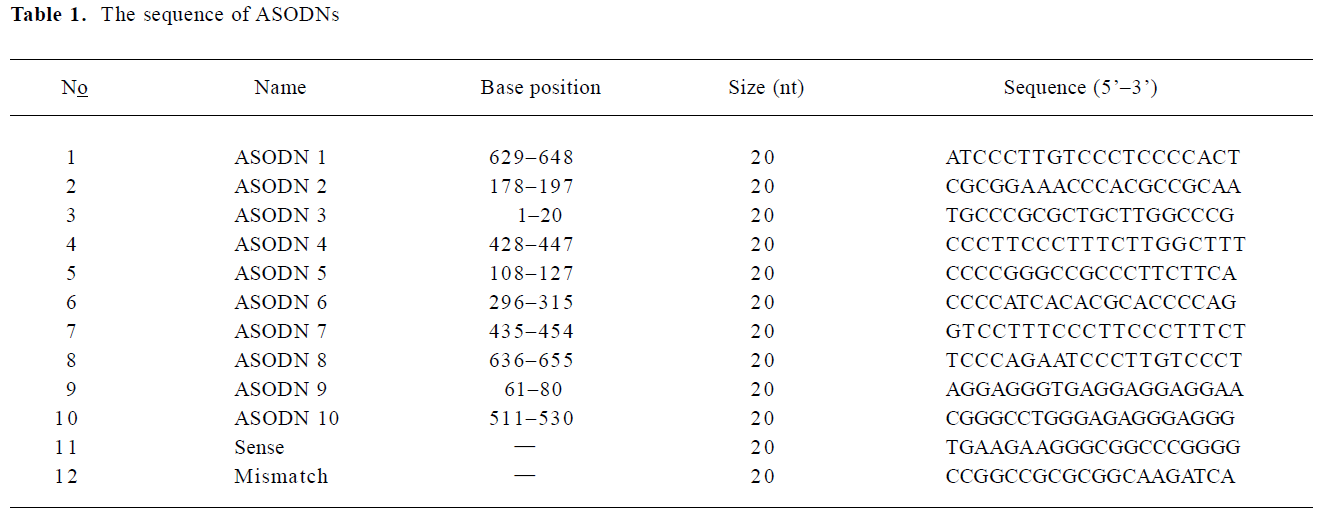
Full table
Cell lines and transfection Human liver hepatocellular carcinoma cell lines (HepG2, SMMC-7721 and BEL-7402 were from the Chinese Academy of Medical Science, Beijing, China) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM)or RPMI-1640 medium (Invitrogen Corporation, CA, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO BRL, Grand Island, NY, USA), 100 U/mL penicillin and 100 µg/mL streptomycin at 37 °C, and 5% CO2. 3×103 cells were seeded in each well of a 96-well microtiter plate and allowed to attach overnight. Oligonucleotides with concentrations of 0.1, 0.2, 0.4, 0.8 µmol/L were transfected into the cells with Lipofectin (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Transfection medium was replaced by normal culture medium after 6 h.
Cell viability assay The effects of ASODN on cellular viability were measured by MTS (3-[4, 5-dimethythiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium, inner salt) assay. After 48 h of incubation following transfection, 20 µL MTS (Sigma, St Louis, MO, USA) was added to each well and incubated at 37 °C for 2 h. The absorbance value was determined at 490 nm by a MR600 Microplate reader (Wallac 1420 Multilable counter, Wallac, Turku, Finland).
To investigate whether the transfection of ASODN enhances chemosensitivity of cancer cells, HepG2 cells were treated by combination of chemotherapeutics (5-fluorouracil (5-FU), adriamycin (ADM) and cis-diaminodichloroplatin(DDP)) and ASODN5 at various concentrations. Cell proliferation was analyzed after 24 h as described earlier. Each assay was performed in quadruplicate. The proliferation inhibition rate was calculated as: (Acontrol–Asample)/(Acontrol–Ablank)×100.
RT-PCR The total RNA from cell lines was extracted using Trizol (Invitrogen, Carlsbad, USA) by a single-step phenol extraction. Subsequent RT-PCR reaction was performed using an RT-PCR kit (Promega, Madison, WI, USA). PCR products were run on 2.0% agarose gel and visualized by ethidium bromide staining. The product intensities were measured by scanning the gel with Gel Doc 1000 (Bio-Rad, Hercules, CA, USA), with GAPDH serving as an internal control.
Western blot analysis The cells in the microplates were collected into 1.5 mL eppendorf tubes in ice and centrifuged at 2000 r/min for 5 min at 4 °C. They were then resuspended in 100 µL cell lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris-Cl, pH 8.0, and 4% SDS), and incubated on ice for 1 h to lyse the cells. The lysates were centrifuged at 14 000 r/min for 15 min at 4 °C; the supernatants were collected and stored at –20 °C for electrophoresis. An extract of each sample was separated on 12% sodium dodecyl sulfate polyacrylamide electrophoretic gels. The separated proteins were blotted onto Hybond-polyvinylidene difluoride membranes (Schleicher & Schuell Biosciences, Inc., Keene N H, USA) and incubated in 5% skimmed milk blocking solution at room temperature overnight. The reactive band was visualized with an ECL-plus Detection Kit (Amersham Biosciences, Piscataway, NJ) and scanned by Gel Doc 1000 (Bio-Rad CA, USA). β-actin was used as a control.
Measurement of apoptosis and caspase-3 activity analysis Apoptosis was assessed for the ASODN5-transfected cells after culture in normal medium for 48 h using Annexin V-FITC/PI (Baosai, Beijing, China) according to the manufacturer’s protocol. Flow cytometry was performed on a FACScalibur machine and the data was analyzed using CellQuest software (SOBR model, Becton Dichinson Instrument, San jose, CA, USA). Caspase-3 activity was measured using the colorimetric protease assay kit according to the manufacturer’s protocol (Calbiochem, La Jolla, CA, USA). HepG2 cells were cultured in 100 mm dishes in DMEM medium plus 10% FBS for 24 h. ASODN5 0.4 µmol/L was added. Cytosol extracts of the cells were diluted to a concentration of 100 µg protein per 50 µL cell lysis buffer. Protease activity was measured using the tetrapeptide p-nitroanilide substrate. The absorbance was measured at 405 nm.
Statistics Data were expressed as mean±SD. Statistical analysis was carried out using Student’s-t-test (two tailed); P<0.05 indicated statistical significance.
Results
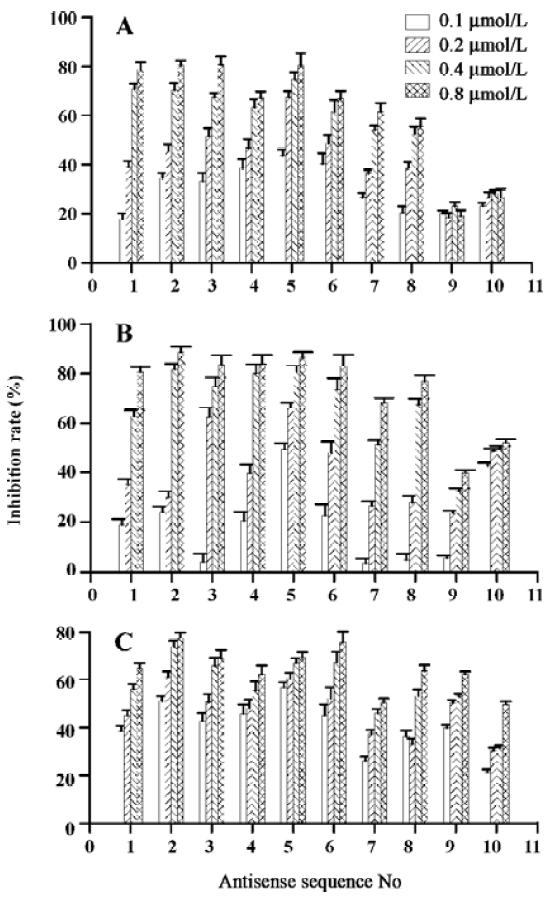
Effects of ASODN on hepatocellular carcinoma cell proliferation The 10 ASODN designed to target against MK were individually screened for their abilities to inhibit hepatocellular carcinoma cell proliferation. All 10 ASODN showed various inhibitory activities on the 3 lines of cancer cells, BEL7402, HepG2, and SMMC-7721. The inhibition rate ranged from 20% to 85%, positively related to the ASODN concentrations (Figure 1). Among the 10 ASODN, ASODN1–6 showed higher inhibitory activity. Meanwhile, we also observed that ASODN6, ASODN5, ASODN3, and ASODN2 had lower IC50, respectively (Figure 2). We therefore choose the 4 ASODN for further experiments.
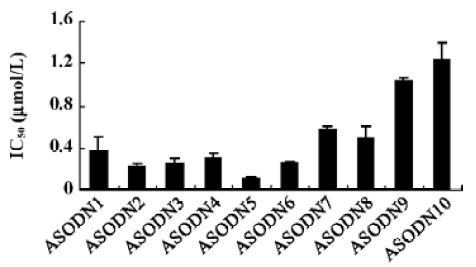
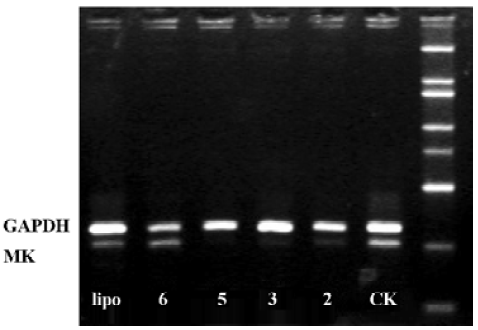
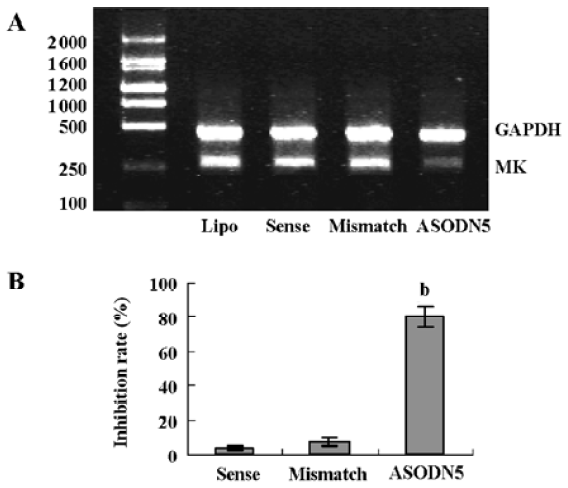
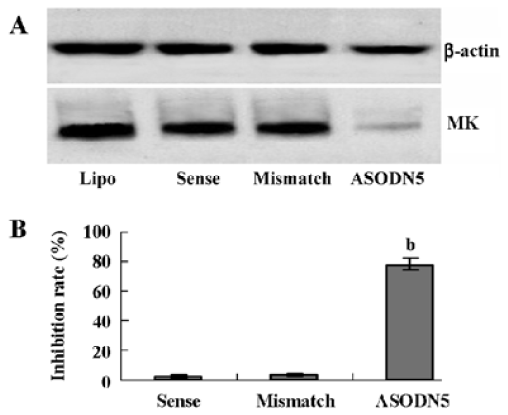
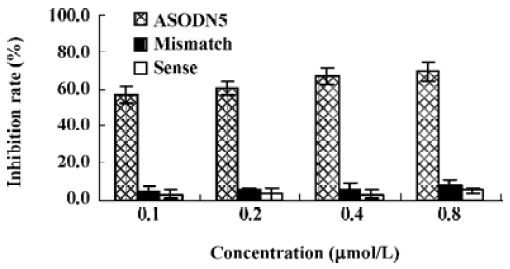
Effects of ASODN on MK expression Since ASODN6, ASODN5, ASODN3 and ASODN2 had lower IC50 respectively, we wanted to determine whether they would decrease MK expression in HepG2 cells. From Figure 3, we obtained that only ASODN5, ASODN3 and ASODN2 downregulated the MK mRNA level significantly in our experiment. Given the lowest IC50 (0.11 µmol/L), we then selected ASODN5 as a candidate for antitumor drug study. To further characterize the function of ASODN5 on MK expression (mRNA and protein), HepG2 cells highly expressing MK were transfected with ASODN5 (0.4 µmol/L), whereas the cells transfected with mismatched compound (0.4 µmol/L), sense compound (0.4 µmol/L) and Lipofectin (0.4 µmol/L) were used as controls. Forty-eight hours after transfection, the cells were examined by RT-PCR and Western blotting. As shown in Figure 4, ASODN5 lowered the MK mRNA level in HepG2 cells, with mismatched, sense compound (0.4 µmol/L) and Lipofectin (0.4 µmol/L) showing no effect. In addition, the MK protein level was decreased by 78.2% (P<0.05) in HepG2 cells by the ASODN5 (Figure 5). Data presented here suggests that ASODN5 can specifically downregulate MK expression. Additionally, mismatched and sense compounds showed little effect on cell proliferation in HepG2 cells (Figure 6).
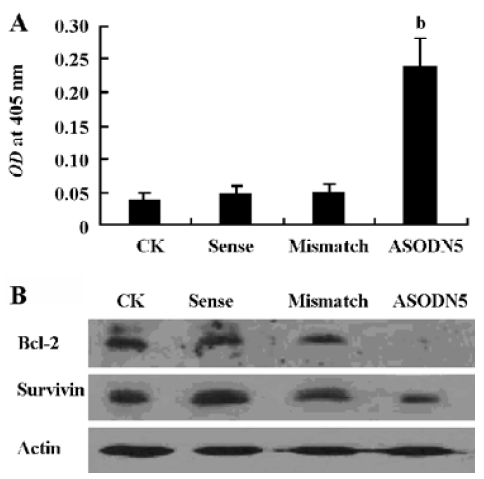
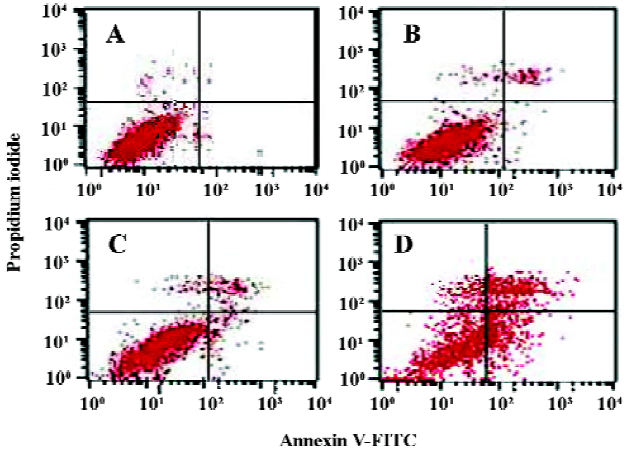
Analysis of cell apoptosis and caspase-3 activity It has been reported that MK protects cells against apoptosis through downregulating caspase-3 activity[35]. In this experiment, the caspase-3 activity increased in the ASODN5-treated cells (Figure 7A). Interestingly, the survivin and Bcl-2 protein content decreased with MK antisense compound transfection in HepG2 cells (Figure 7B). Since the ASODN5 can downregulate caspase-3 activity, then flow cytometry was then used to quantify the changes in cell apoptosis after transfection with ASODN5 for 48 h. From Figure 8, it was clear that there was about 38.0%±2.5% (n=3) cell entry apoptosis after transfection with ASODN5.
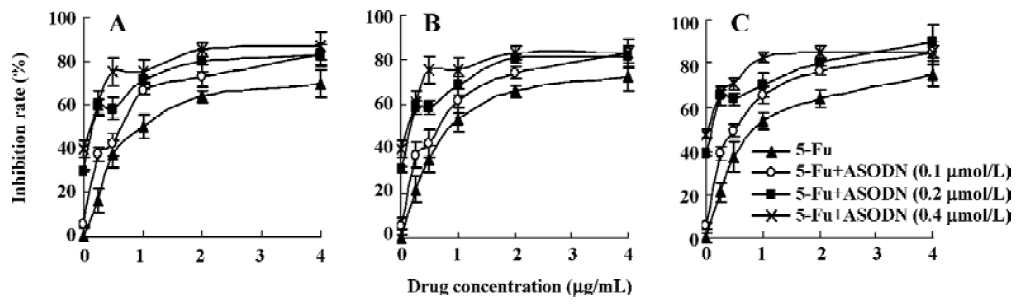
Inhibition of cell proliferation after combination with chemotherapeutic drugs Figure 9 shows the results of combination therapy with chemotherapeutic drugs. It is clear that chemotherapeutic drugs 5-Fu, ADM and DDP can significantly reduce HepG2 cell growth, and the inhibitory effect exhibited a dose-dependent manner. It is interesting to observe that ASODN5 transfer can increase the chemosensitivity of 5-Fu, ADM and DDP. It is noted that the synergistic cytotoxic effect is more effective at low concentrations.
Discussion
Accumulating data suggest that MK, a heparin-binding growth factor, is overexpressed in a wide range of human carcinomas and is believed to contribute to tumorigenesis and tumor progression. Antisense approaches are efficient tools for the inhibition of gene expression in a sequence-specific way. However, it is known that the screen of high active ASODN is difficult because of the complexity of the RNA structure[35]. In the present study, we generated sequence-specific antisense compounds to downregulate MK expression (mRNA and protein) in hepatocellular carcinoma cells leading to the inhibition of their growth.
In this study, we first designed antisense sequences using a neural network tool, and several candidate sequences were obtained. In order to improve the effectiveness of ASODN, the MK mRNA secondary structure was also predicted according to Zuker[36]. Using this theoretical approach, 10 putative target sites were identified (Table 1). Our data show that all of the 10 ASODN have inhibitory activity, and ASODN5 has the lowest IC50 at 0.11 µmol/L. The result suggests that the computational method is a good approach for finding putative target sites.
As measured by RT-PCR and Western blotting, the antisense compounds efficiently downregulated the MK expression level in HepG2 cells in a dose-dependent manner. However, the transfection of mismatched and sense compounds showed little effect on MK expression in HepG2 cells (Figures 4,5). At the same time, ASODN5 transfection inhibited the proliferation of HepG2 cells significantly. Taken together, our data suggests that the proliferation inhibition by ASODN was due to the downregulation of MK expression.
Several mechanisms were proposed to increase cell proliferation by MK. Among these, the downregulation of caspase-3 activity is well recognized. Here the antisense compounds induced a strong growth-inhibitory effect and apoptosis on HepG2 cells. We also found that caspase-3 protease activity increased in cells transfected with antisense targeting MK (Figure 7A). Bcl-2 and the survivin protein were thought to be very important anti-apoptotic proteins in cells. Survivin can interact with the caspase-3 enzyme and lead to a reduction of caspase-3 enzyme activity[37]. Addi-tionally, Bcl-2 can form a complex with Bax, and then suppress the activation of caspase-3[38]. Since MK antisense transfection can downregulate caspase-3 activity, we wanted to determine whether the declined caspase-3 activity is associated with survivin and Bcl-2. To our surprise, MK antisense transfection led to a marked decline of survivin and Bcl-2 protein expression (Figure 7B). We therefore speculated that the downregulation of survivin and Bcl-2 protein expression by MK antisense transfection in one of the important ways to activate caspase-3 activity, subsequently inducing cell apoptosis. Our data presented here also provide new anti-apoptotic mechanisms of MK. In short, these data suggest that the loss of MK expression is sufficient to trigger caspase-dependent apoptosis in HepG2 cells.
Drug tolerance is often developed during combination chemotherapy of HCC. Thus, the approach of enhancing chemosensitivity by using apoptosis-inducing agents appears to be a proper strategy for patients with advanced cancers. It is known that MK is overexpressed in liver hepatocellular cells and can restrain cell apoptosis through the downregulation of caspase-3 activity. Therefore, MK antisense transfer is anticipated to increase chemosensitivity. In our work, 3 chemotherapeutics (5-Fu, ADM and DDP) combined with ASODN5 targeting MK, increased chemosensitivity significantly. Downregulation of MK could be a promising strategy to reduce drug resistance of hepatocellular carcinoma.
References
- Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun 1988;151:1312-8.
- Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, et al. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science 1990;250:1690-4.
- Merenmies J, Rauvala H. Molecular cloning of the 18-kDa growth-associated protein of developing brain. J Biol Chem 1990;265:16721-4.
- Muramatsu T. Midkine (MK), the product of a retinoic acid responsive gene, and pleiotrophin constitute a new protein family regulating growth and differentiation. Int J Dev Biol 1993;37:183-8.
- Garver RI, Chan CS, Milner PG. Reciprocal expression of pleiotrophin and midkine in normal versus malignant lung tissues. Am J Respir Cell Mol Biol 1993;9:463-6.
- Garver RI, Radford DM, Donis-Keller H, Wick MR, Milner PG. Midkine and pleiotrophin expression in normal and malignant breast tissue. Cancer 1994;74:1584-90.
- Aridome K, Tsutsui J, Takao S, Kadomatsu K, Ozawa M, Aikou T. Increased midkine gene expression in human gastrointestinal cancers. Jpn J Cancer Res 1995;86:655-61.
- Nakanishi T, Kadomatsu K, Okamoto T, Tomoda Y, Muramatsu T. Expression of midkine and pleiotropin in ovarian tumors. Obstet Gynecol 1997;90:285-90.
- O’Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. The angiogenic factor midkine is expressed in bladder cancer, and overexpression correlates with a poor outcome in patients with invasive cancers. Cancer Res 1996;56:2515-8.
- Konishi N, Nakamura M, Nakaoka S, Hiasa Y, Cho M, Uemura H, et al. Immuno-histochemical analysis of midkine expression in human prostate carcinoma. Oncology 1999;57:253-7.
- Mishima K, Asai A, Kadomatsu K, Ino Y, Nomura K, Narita Y, et al. Increased expression of midkine during the progression of human astrocytomas. Neurosci Lett 1997;233:29-32.
- Nakagawara A, Milbrandt J, Muramatsu T, Deuel TF, Zhao H, Cnaan A, et al. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer Res 1995;55:1792-7.
- Tsutsui J, Kadomatsu K, Matsubara S, Nakagawara A, Hamanoue M, Takao S, et al. A new family of heparin-binding growth/differentiation factors: increased midkine expression in Wilms’ tumor and other human carcinomas. Cancer Res 1993;53:1281-5.
- Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, et al. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science 1990;250:1690-4.
- Shimada H, Nabeya Y, Tagawa M, Okazumi S, Matsubara H, Okazumi S, et al. Preoperative serum midkine concentration is a prognostic marker for esophageal squamous cell carcinoma. Cancer Sci 2003;94:628-32.
- Ikematsu S, Nakagawara A, Nakamura Y, Sakuma S, Wakai K, Muramatsu T, et al. Correlation of elevated level of blood midkine with poor prognostic factors of human neuroblastomas. Br J Cancer 2003;88:1522-6.
- Obata Y, Kikuchi S, Lin Y, Yagyu K, Muramatsu T, Kumai H, et al. Serum midkine concentrations and gastric cancer. Cancer Sci 2005;96:54-6.
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, et al. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem 1999;73:2084-92.
- Qi M, Ikematsu S, Ichihara-Tanaka K, Sakuma S, Muramatsu T, Kadomatsu K. Midkine rescues Wilms’ tumor cells from cisplatin-induced apoptosis: regulation of Bcl-2 expression by midkine. J Biochem 2000;127:269-77.
- Muramatsu H, Muramatsu T. Purification of recombinant midkine and examination of its biological activities: functional comparison of new heparin binding factors. Biochem Biophys Res Commun 1991;177:652-8.
- Muramatsu H, Shirahama H, Yonezawa S, Maruta H, Muramatsu T. Midkine (MK), a retinoic acid-inducible growth/differentiation factor: immunochemical evidence for the function and distribution. Dev Biol 1993;159:392-402.
- Takada T, Toriyama K, Muramatsu H, Song XJ, Torii S, Muramatsu T. Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J Biochem 1997;122:453-8.
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J Biol Chem 1999; 274: 12 474–9.
- Horiba M, Kadomatsu K, Nakamura E, Muramatsu H, Ikematsu S, Sakuma S, et al. Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J Clin Invest 2000;105:489-95.
- Sandra F, Harada H, Nakamura N, Ohishi M. Midkine induced growth of ameloblastoma through MAPK and Akt pathways. Oral Oncol 2004;40:274-80.
- Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991;350:427-8.
- Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 1998;4:1065-7.
- Thorgeirsson SS, Teramoto T, Factor VM. Dysregulation of apoptosis in hepatocellular carcinoma. Semin Liver Dis 1998;18:115-22.
- Lau SH, Guan XY. Cytogenetic and molecular genetic alterations in hepatocellular carcinoma. Acta Pharmacol Sin 2005;26:659-65.
- Park YN, Chae KJ, Kim YB, Park C, Theise N. Apoptosis and proliferation in hepatocarcinogenesis related to cirrhosis. Cancer 2001;92:2733-8.
- Kato M, Shinozawa T, Kato S, Awaya A, Terada T. Increased midkine expression in hepatocellular carcinoma. Arch Pathol Lab Med 2000;124:848-52.
- Dai JM, Zhang SQ, Zhang W, Lin RX, Ji ZZ, Wang SQ. Antisense oligodeoxynucleotides targeting the serine/threonine kinase Pim-2 inhibited proliferation of DU-145 cells. Acta Pharmacol Sin 2005;26:364-8.
- Chalk AM, Sonnhammer EL. Computational antisense oligo prediction with a neural network model. Bioinformatics 2002;18:1567-75.
- Ohuchida T, Okamoto K, Akahane K, Higure A, Todoroki H, Abe Y, et al. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer 2004;100:2430-6.
- Jiang QS, Wang SQ. Design and screening of antisense oligodeoxynucleotides against PAI-1mRNA in endothelial cells in vitro. Acta Pharmacol Sin 2006;27:1018-23.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003;31:3406-15.
- Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry 2001;40:1117-23.
- Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 1997;385:637-40.