
Involvement of mitochondria and caspase pathways in N-demethyl-clarithromycin-induced apoptosis in human cervical cancer HeLa cell
Introduction
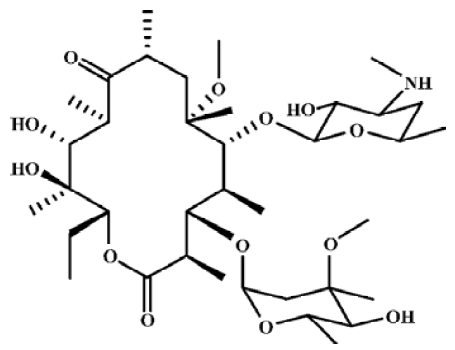
Macrolide antibiotics are widely used as antibacterial and anti-inflammatory agents. Macrolides have been used successfully to treat diffuse panbronchiolitis –– a progressive inflammatory lung disease, and might be useful for the treatment of asthma, chronic bronchitis, chronic sinusitis, cystic fibrosis and bronchiectasis. In vivo and in vitro studies show that macrolides affect neutrophil chemotaxis and infiltration, inflammatory cytokine production, mucus production and the transportability of airway secretions[1]. Some studies have also focused on their anti-tumor activities[2]. N-demethyl-clarithromycin (Figure 1) is the main metabolite of clarithromycin in vivo, and it had been reported that N-demethyl-clarithromycin has potent anti-inflammatory activity[3], inhibits the proliferation of T lymphocyte and induces a cell cycle arrest at the G2/M stage in inflammatory cells, however, there has been no report on its anticancer activity. Therefore, the apoptotic effect of N-demethyl-clarithromycin on human cervical cancer HeLa cells was investigated for the first time in this study.
Apoptosis, or programmed cell death, is an important process in biological systems, including normal cell turnover, immune system, embryonic development, metamorphosis and endocrine-dependent tissue atrophy. Cells undergoing apoptosis execute the death program by activating caspases. Caspase, a family of at least 14 related cysteine proteases, are mediators of apoptotic processes caused by various inducers such as Fas ligand, TNFα, etoposide and actinomycin D[4–7].
It has been shown that the mitochondrial Bcl-2 protein family plays an essential role in apoptosis. This family is a large group of apoptosis-regulating proteins that modulate mitochondrial pathways. It includes both anti-apoptotic proteins, (eg Bcl-2, Bcl-XL, and Mcl-1) and various pro-apoptotic proteins (eg Bax, Bak, and Bad)[8]. These proteins regulate mitochondrial membrane permeability, either promoting or suppressing the release of apoptogenic proteins from these organelles[9].
The tumor suppressor p53 plays an important role in regulating the expression of genes that mediate cell cycle arrest and/or apoptosis in response to genotoxic insults[10]. SIRT1 is a key regulator involved in cell aging and the response to DNA damage[11], and it is a member of the sirtuin family of nicotinamine adenine dinucleotide (NAD+)-dependent deacetylase and known to inhibit stress-induced apoptosis.
In the present study, we investigated the mechanism of N-demethyl-clarithromycin-induced HeLa cell apoptosis through the mitochondrial pathway.
Materials and methods
Chemical reagents N-demethyl-clarithromycin was synthesized by the demethylation of clarithromycin in the presence of iodine. Its purity was about 98%, determined by HPLC method. It was dissolved in DMSO; the concentration of DMSO in all cell cultures was kept at less than 0.1%, which had no detectable effect on cell growth or apoptosis.
Fetal bovine serum (FBS) was purchased from TBD Biotechnology Development (Tianjin, China); propidium iodide (PI) and RNase A were purchased from Sigma Chemical (St Louis, MO, USA); thiazolyl blue (MTT) was obtained from Sino-American Biotechnology (Beijing, China); pan-caspase inhibitor (z-VAD-fmk), caspase-3 inhibitor (z-DEVD-fmk), caspase-9 inhibitor (z-LEHD-fmk) and caspase-8 inhibitor (z-IETD-fmk) were purchased from Enzyme Systems (Liver-more, CA, USA); rabbit polyclonal antibodies against Bax, caspase-activated DNase (ICAD), cytochrome c, p53, mouse polyclonal antibodies against Bcl-2 and horseradish peroxidase-conjugated secondary antibody (goat-anti-rabbit or goat-anti-mouse) were purchased from Santa Cruz Biotechnology (Santa Cruz, USA).
Cell culture The cervical carcinoma HeLa cell line was obtained from American Type Culture Collection (ATCC, #CCL-2, Rochville, MD, USA). The cells were cultured in RPMI-1640 medium (GIBCO, Grand Island, NY, USA) supplemented with 10% FBS and 0.03% L-glutamine (GIBCO,USA) and maintained at 37 ºC with 5% CO2 in a humidified atmosphere.
Cytotoxicity assay The cytotoxic effect of N-demethyl-clarithromycin on HeLa cells was measured by MTT assay as described[12]. All the cells were cultured at 5×104 cells/well in 96-well plates (NUNC, Roskilde, Denmark) without or with z-VAD-fmk, z-DEVD-fmk, z-LEHD-fmk and z-IETD-fmk at given concentrations for 1 h, and then treated with N-demethyl-clarithromycin. Four hours before the end of incubation, 20 µL MTT solutions (5.0 mg/L) were added to each well. Resulting crystals were dissolved in DMSO. The result was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazo-lium (MTT) assay using a plate microreader (TECAN SPECTRA, Wetzlar, Germany). The percentage of cell viability was calculated as follows:
Cell viability (%)=A492 (drug)/A492 (control)×100 (1)
Observation of morphologic changes HeLa cells in RPMI- 1640 containing 10% FBS were seeded into 6-well culture plates and cultured for 24 h before 60 µmol/L N-demethyl-clarithromycin treatment. The cellular morphology was observed using a phase-contrast microscopy (Leica, Wetzlar, Germany) at 0, 12, 24, 36, or 48 h.
Nuclear damage observed by Hoechst 33258 staining The cells in RPMI-1640 containing 10% FBS were seeded into 6-well plates and incubated overnight. N-demethyl-clarithromycin was added to the cell culture and incubated for the indicated times. The cells were fixed with 3.7% parafor-maldelyde at room temperature for 2 h, then washed twice with Ca2+ and Mg2+ free phosphate-buffered saline (PBS) and stained with Hoechst 33258 (Sigma, USA) 167 µmol/L at 37 ºC for 15 min. At the end of incubation, the nuclear morphology of the cells was observed under a fluorescence microscope (Olympus-AX70, Tokyo, Japan).
DNA extraction and detection of DNA fragments DNA extraction and electrophoresis were performed as described previously[13]. In brief, HeLa cells, both adherent and floating, were collected by centrifugation at 1000×g for 5 min. The cell pellet was suspended in cell lysis buffer (Tris-HCl 10 mmol//L pH 7.4, edetic acid 10 mmol/L pH 8.0 and Triton-X 100 0.5%) and kept at 4 ºC for 30 min. The lysate was centrifuged at 25 000×g for 20 min. The supernatant was incubated with RNase A 20 µg/L at 37 ºC for 1 h, and then incubated with proteinase K 20 µg/L at 37 ºC for 1 h. The resulting solution was mixed with NaCl 5 mol/L and dimethylcarbinol at -20 ºC overnight, and then centrifuged at 25 000×g for 15 min. After drying, the DNA was dissolved in TE (Tris-HCl 10 mmol/L, pH 7.5) buffer and separated by 2% agarose gel electrophoresis at 100 V and ethidium bromide staining.
Lactate dehydrogenase activity-based cytotoxicity assays The cells were cultured with N-demethyl-clarithromycin for 0, 12, 24, 36, or 48 h. Floating cells were collected from culture medium by centrifugation (240×g) at 4 ºC for 5 min, and the lactate dehydrogenase (LDH) content from the pellets lysed in 1% NP-40 for 15 min was used as an index of apoptotic cell death (LDHp)[14]. The LDH released in the culture supernatant (extracellular LDH or LDHe) was used as an index of necrotic death, and the LDH present in the adherent viable cells as intracellular LDH (LDHi). Then the substrate reaction buffer of LDH [L (+)-lactic acid 0.5 mmol/L, indonitro-tetrazolium 0.66 mmol/L, phenazine methosulfate 0.28 mmol/L, nicotinamide adenine dinucleotide 1.3 mmol/L in pH 8.2 Tris-HCl] was added. The optical density (OD) value at 492 nm of the reaction for 1 and 5 min were assayed and LDH activities were determined by the average difference between 1 and 5 min. The percentage of apoptotic and necrotic cell death was calculated as follows:
apoptosis%=LDHp/(LDHp+LDHi+LDHe)×100
necrosis%=LDHe/(LDHp+LDHi+LDHe)×100 (2)
Measurement of mitochondrial transmembrane potential (ΔΨm) To measure the mitochondrial transmembrane potential (ΔΨm), the cells were treated with 60 µmol/L N-demethyl-clarithromycin at the indicated time, washed with PBS, and then incubated with PBS containing 1 µg/mL rhodamine 123 at room temperature for 30 min. After 2 washes and final resuspension in PBS, the fluorescence of cells was observed using fluorescence microscopy (Leica, Wetzlar, Germany) and measured by a FACScan flowcytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis HeLa cells were treated with 60 µmol/L N-demethyl-clarithromycin for 0, 12, 24, 36, or 48 h. Both adherent and floating cells were collected and Western blot analysis was performed as previously described[15]. Briefly, the cells were washed with ice-cold PBS and solubilized with lysis buffer (1% SDS, 100 mmol/L sodium fluoride, 50 mmol/L HEPES (pH 7.4), 1% Triton X-100, 1 mmol/L phenylmethyl-sulfonylfluoride (PMSF), 1 mmol/L EDTA, 2 mmol/L leupeptin and 1 mmol/L aprotinin). Protein concentration was determined by the Bio-Rad DC protein assay (Bio-Rad Laboratories, Bio-rad, Richmond, CA). The protein lysates were separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were soaked in blocking buffer (5% skimmed milk), and then incubated with primary antibodies, followed by horseradish peroxidase conjugated secondary antibodies overnight. The color was developed with diaminobenzidine.
Preparation of mitochondrial and cytosolic extracts After treatment with or without N-demethyl-clarithromycin, the cells were collected by centrifugation at 200×g at 4 °C for 5 min and then washed twice with ice-cold PBS. The cell pellets were resuspended in ice-cold homogenizing buffer, including 250 nmol/L sucrose, 20 mmol/L HEPES, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1.5 mmol/L MgCl2, 1 mmol/L DTT, 1 mmol/L PMSF, 1 µg/mL aprotinin and l µg/mL leupeptin. After homogenization (40 strokes), the homo-genates were centrifuged at 4200×g at 4 °C for 30 min. The supernatants and the pellets were analyzed by Western blotting, respectively. The supernatant was used as the cytosol fraction, and the pellet was resolved in lysis buffer as the membrane fraction[16].
Statistical analysis All data and results presented were confirmed in at least 3 independent experiments. The data were expressed as mean±SD. Statistical comparisons were made by Student’s t-test. P<0.05 was considered significant.
Results
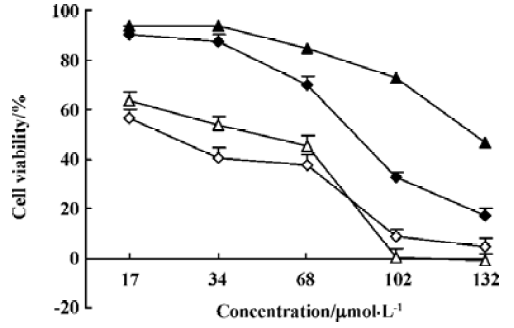
Cytotoxic effects of N-demethyl-clarithromycin on HeLa cells To investigate the cytotoxic effects of N-demethyl-clarithromycin against HeLa cells, the cells were cultured with N-demethyl-clarithromycin at various doses at the indicated times. As shown in Figure 2, N-demethyl-clarithro-mycin inhibited HeLa cell growth in a dose- and time-dependent manner.
N-demethyl-clarithromycin-induced apoptotic cell death in HeLa cells To determine the characteristics of N-demethyl-clarithromycin-induced HeLa cell death, morphologic changes and DNA fragmentation were examined. In HeLa cells, exposure to N-demethyl-clarithromycin 60 µmol/L for 24 h resulted in morphologic alterations characteristic of apoptosis, including membrane blebbing, nuclear condensation and granular apoptotic bodies (Figure 3C, 3G), compared with the control group (Figure 3A). In gel electro-phoresis, DNA fragmentation as a hallmark of apoptosis was observed in N-demethyl-clarithromycin treated cells (Figure 3H).
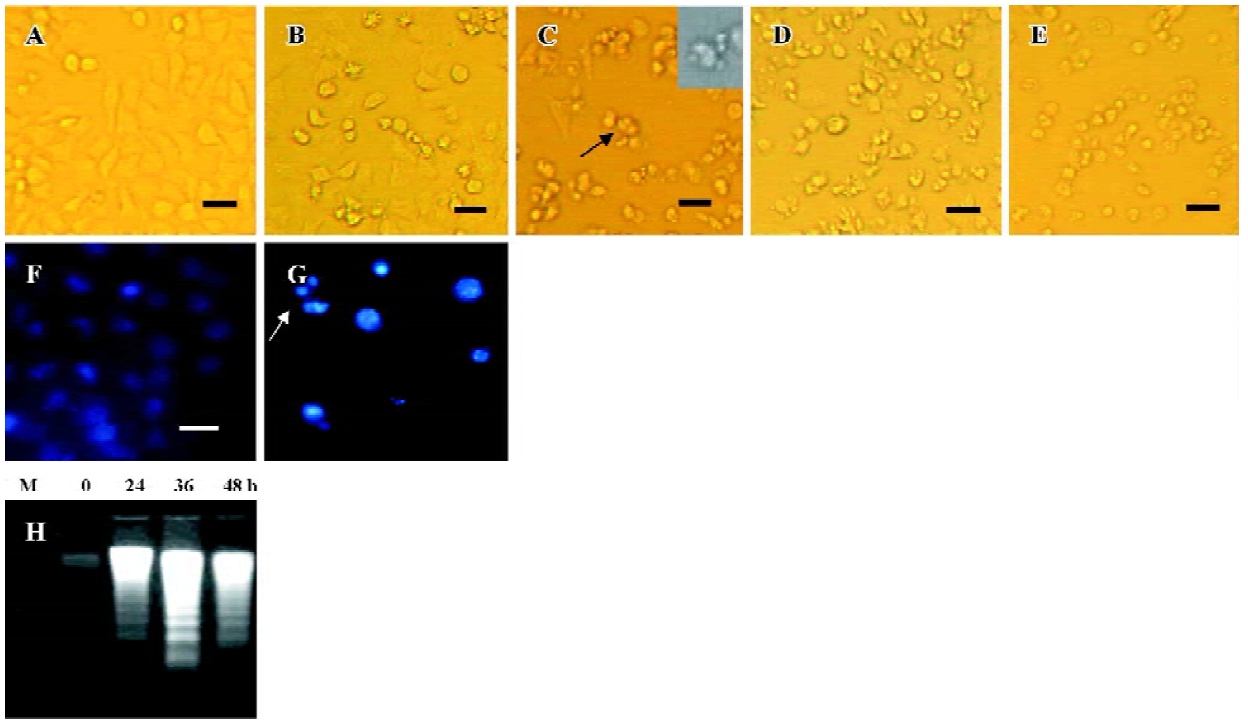
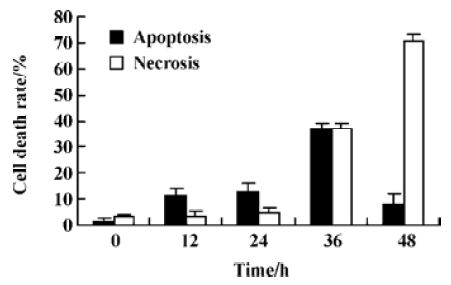
Characterization of HeLa cell death identified by LDH release assay In order to further investigate whether N-demethyl-clarithromycin also initiated necrosis in HeLa cells, the rates of LDH released from viable cells, floating dead cell and the culture medium were measured. The percentage of apoptosis increased from 11.18% to 37.30% in a time-dependent manner. It reached the peak at 36 h, then began to decrease, while the percentage of necrosis continued to increase from 3.30% to 70.44% (Figure 4), and similar results were observed in morphologic changes (Figure 3B–3E).
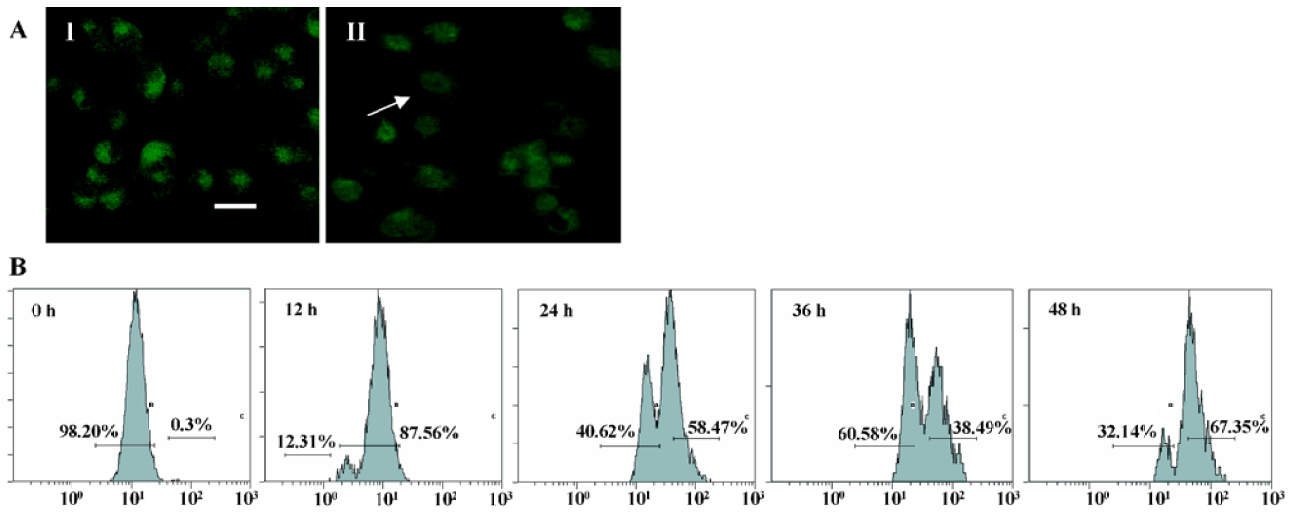
Mitochondrial transmembrane potential (ΔΨm) was markedly reduced by N-demethyl-clarithromycin treatment Kinetic data indicate that mitochondria undergo major changes in membrane integrity before classical signs of apoptosis become manifest[17]. These changes concern both the inner and the outer mitochondrial membranes, leading to a disruption of the inner transmembrane potential (ΔΨm) and the release of intermembrane proteins through the outer membrane. In this study, mitochondrial transmembrane potential (ΔΨm) was further assessed using rhodamine 123, a specific fluorescent probe for the analysis of mitochondrial transmembrane potential. Fluorescent intensity was significantly decreased in N-demethyl-clarithromycin treated cells (Figure 5A II), compared with the control group (Figure 5A, I). The mitochondrial transmembrane potential was decreased in a time-dependent manner, and the major decrease occurred at 36 h (Figure 5B), suggesting that N-demethyl-clarithro-mycin induced HeLa cell apoptosis through mitochondrial pathway.
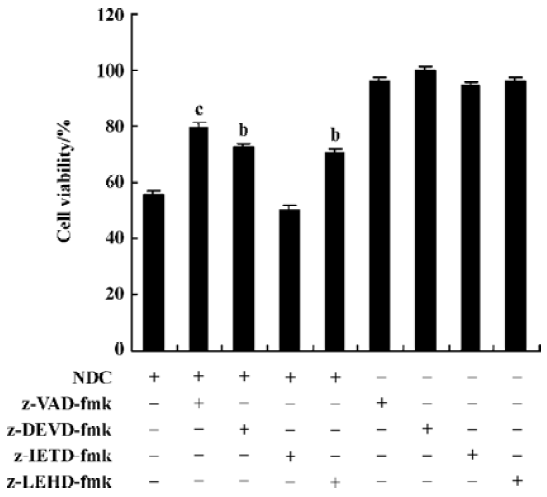
Caspase-3 was activated during N-demethyl-clarithro-mycin induced-HeLa cell death The pan-caspase inhibitor (z-VAD-fmk), the caspase-3 inhibitor (z-DEVD-fmk), the caspase-9 inhibitor (z-LEHD-fmk) 20 µmol/L, effectively enhance cell viability induced by N-demethyl-clarithromycin, but the caspase-8 inhibitor (z-IETD-fmk) had almost no effect on HeLa cell viability (Figure 6).
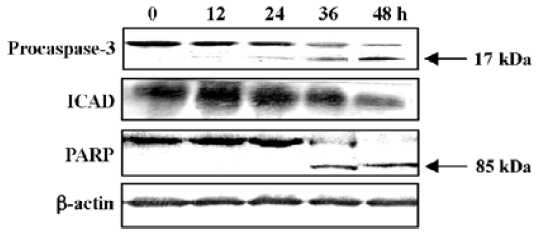
Western blot analysis was carried out to further confirm the participation of caspase-3. The 32-kDa band of proca-spase-3 was degraded after treatment with N-demethyl-clarithromycin for 24 h, indicating the activation of caspase-3. The inhibitor of a caspase-dependent DNase, referred to as ICAD (DEF40/CPAN), is a caspase-3 substrate that is cleaved to be inactivated and allow caspase-dependent DNase to execute the characteristic fragmentation of DNA. ICAD is expressed as 2 isoforms: one is 45 kDa (ICAD-L/DFF45) and the other is 35 kDa (ICAD/DFF35). Although both ICAD-L/DFF45 and ICAD/DFF35 bind to and inhibit CAD, only ICAD-L/DFF45 was reported to be functional[18,19]. Thus, the ICAD-L/DFF45 expression in HeLa cells was examined to confirm the activation of caspase-3. After exposure to N-demethyl-clarithromycin 60 µmol/L for 12 h, ICAD-L was degraded. Another characteristic associated with the execution phase of the apoptosis pathway is the specific poly-(ADP-ribose) polymerase (PARP) cleavage by the caspase. This cleavage leads to inactivation of the enzyme, thus preventing futile DNA repair cycles[20]. It has been reported that caspase-3 is the most efficient processing enzyme for PARP[21]. Cleavage of PARP was examined after treatment with N-demethyl-clarithromycin. As expected, the amount of 116 kDa protein decreased and the 85 kDa degraded product increased after 36 h (Figure 7).
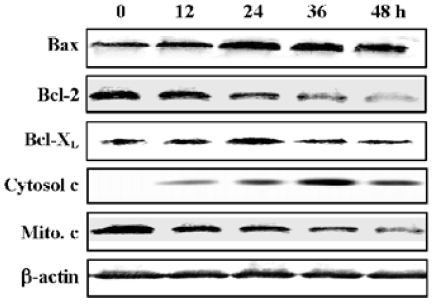
Release of cytochrome c and increased Bax/Bcl-2 expression ratio in N-demethyl-clarithromycin-treated HeLa cells Since Bcl-2 family members are critical regulators of the mitochondrial pathway that induces intrinsic activation of caspase, Western blot analysis was carried out to observe the expression of the Bcl-2 family proteins Bcl-2, Bcl-XL, Bax and the release of cytochrome c. Twelve hours after treatment with N-demethyl-clarithromycin, the expression of pro-apoptotic Bax and the release of cytochrome c in the cytosol began to increase, whereas that of anti-apoptotic Bcl-2 and cytochrome c in the mitochondria began to decrease slightly, and Bcl-XL almost had no change. These results suggest that N-demethyl-clarithromycin-induced apoptosis involved the initial phase mediated by the balance between Bcl-2 and Bax, resulting in cytochrome c release from the mitochondria (Figure 8).
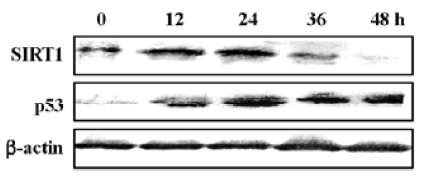
SIRT1 expression was downregulated in N-demethyl-clarithromycin-induced HeLa cell death The inactivation of the SIRT1 protein can potentiate p53- or Bax-mediated apoptosis[11,22]. Western blot analysis was carried out to observe the expression of SIRT1 and p53 in N-demethyl-clarithromycin-treated HeLa cell death. As shown in Figure 9, SIRT1 protein expression began to decrease after incubation with N-demethyl-clarithromycin for 36 h, while p53 expression was upregulated in 12 h.
Discussion
This study demonstrates that N-demethyl-clarithromycin inhibits the proliferation of HeLa cell, and based on morphologic observation, DNA fragmentation suggests that N-demethyl-clarithromycin-induced HeLa cell death involved a mechanism of apoptosis. In the LDH assay, N-demethyl-clarithromycin-induced HeLa cell apoptosis was the predominant mechanism of cell death before 36 h; after prolonged exposure to N-demethyl-clarithromycin at the time, necrosis was also obvious, suggesting that necrosis played an important role after 36 h and apoptosis also could orchestrate necrosis. Therefore, we conclude that N-demethyl-clarithromycin induced HeLa cell death through the balance between apoptosis and necrosis. Morphological changes further confirmed that marked nuclear fragmentation was observed in a time-dependent manner.
Caspases are a family of cysteases, which cleave protein substrates after their Asp residues; caspases appear to be involved in regulating the activation of apoptotic signal transmission. The result of this study showed that pretreatment with the caspase inhibitors z-VAD-fmk, z-DEVD-fmk, and z-LEHD-fmk effectively enhanced N-demethyl-clarithromycin-induced cell viability, but the caspase-8 inhibitor (z-IETD-fmk) had almost no effect on HeLa cell viability. Whether some death receptors participated in the event or not should be further investigated. To further confirm the participation of caspase-3 in the cell death, we examined the protein expression of caspase-3, ICAD, and PARP. The expression of procaspase 3 was downregulated and the protein expressions of ICAD and PARP expression were significantly downregulated. All these results suggest that the caspase cascade played a critical role in N-demethylclarithro-mycin-mediated HeLa cell apoptosis.
Several pro-apoptotic proteins, such as Bax, Bak, and Bid, translocate to the mitochondrial membrane, and this localization is associated with their pro-apoptotic activities. It has been reported that Bcl-2 exerts its action through heterodimerization with Bax. Meanwhile, Bcl-2 and Bcl-XL are cleaved by caspase-3 and are converted to pro-apoptotic proteins similar to Bax. Therefore, the ratio between Bcl-2 and Bax or Bcl-XL and Bax is a decisive factor to activate cell death[23–26]. The oligomerization of Bax in the mitochondrial membrane was shown to induce cytochrome c release, meanwhile pro-apoptotic Bcl-2 cleavage product localized on the mitochondrial membrane and caused a release of cytochrome c[27,28]. The release of cytochrome c from mitochondria to cytosol is an important step in the apoptotic pathway. Such release leads to a caspase-9-dependent activation of caspase-3, as well as a cleaved form of PARP. In this study, HeLa cells treated with N-demethyl-clarithromycin exhibited the elevated ratio between pro-apoptotic Bax and anti-apoptotic Bcl-2 or Bcl-XL. The release of cytochrome c in the cytosol was markedly upregulated, followed by the changes of Bcl-2/Bax and Bcl-XL/Bax ratios at 12 h. Cytochrome c in the mitochondrion was downregulated slightly, suggesting that cytochrome c was released into the cytosol from the mitochondrion. Moreover, the mitochondrial transmembrane potential also decreased. The caspase-9 inhibitor protected the cells from N-demethyl-clarithromycin-induced cell death, and caspase-3 activation was followed by the degradation of the caspase-3 substrates. These results suggest that the mitochondrial pathway of cell death, including the Bcl-2 family and the release of cytochrome c, might be involved in and orchestrate the caspase cascades.
Following environmental insults, p53 is activated by post-translational modifications such as phosphorylation and acetylation which increase its protein stability and enhance its DNA binding activity[29]. Activated p53 upregulates the expression of downstream target genes, including Bax, the p53-upregulated modulator of apoptosis[30]. As Bax expression increased, our attention was drawn to p53 because of the existence of the p53-binding domain in the Bax gene-promoter region. Several studies have indicated that p53 activates the apoptotic machinery through the regulation of the Bax function and mitochondrial integrity, which results in the release of cytochrome c and the activation of caspases. Although HeLa cells express low levels of p53 owing to being infected by HPV18 to block p53 activity, some studies have reported that treatment with drugs could increase p53 expression by inhibition of E6 activity or lead to a persistent increase of p53, and also could accumulate p53 in the nuclei etc[31,32]. In our study, N-demethyl-clarithromycin induced rapid accumulation of p53 at 12 h, which was positively correlated with subsequent upregulation of Bax and resulting activation of caspase-3. This indicates that p53 partly plays a role in the initiation phase of N-demethyl-clarithromycin-induced HeLa cell apoptosis; the more detail mechanism should be further investigated.
SIRT1 is known to inhibit stress-induced apoptosis. SIRT1 regulates p53 function via deacetylation with a specificity for p53 C-terminal Lys382 residue, modification of which has been implicated in the activation of p53 as a transcription factor[22]. It also inhibits stress-induced apoptotic cell death by de-acetylating the DNA repair factor Ku70 that is tightly associated with Bax, causing sequestering Bax away from mitochondria[33]. In this study, N-demethyl-clarithromycin reduced SIRT1 expression in 36 h, while the level of p53 and Bax changed before that of SIRT1, so it implied that other regulators might contribute to the modulation. Taken all our data together, N-demethyl-clarithromycin activates the mitochondria-mediated apoptotic pathway including cytochrome c release and subsequent activation of caspase-9 and caspase-3. These results indicate that N-demethyl-clarithromycin-induced apoptosis of HeLa cells may involve a mitochondrial-related caspase pathway.
Furthermore, other pathways regulating apoptosis, such as c-Jun N-terminal kinase (JNK) and p38 pathways, were not examined in this study and therefore the impact of N-demethyl-clarithromycin on these pathways could not be ruled out and should be further investigated.
References
- Keicho N, Kudoh S, Yotsumoto H. Erythromycin promotes monocyte to macrophage differentiation. J Antibiot 1994;46:80-9.
- Williams JD. Non-antimicrobial activities of macrolides. Int J Antimicrob Agents 2001;18:S89-91.
- Ohmori S, Ishii I, Kuriya S, Taniguchi T, Rikihisa T, Hirose S, et al. Effect of clarithromycin and its metabolites on the mixed-function oxidase system in hepatic microsomes of rats. Drug Metab Dispos 1993;21:358-63.
- Kim SO, Han J. Pan-caspase inhibitor zVAD enhances cell death in RAW 246.7 macrophages. J Endotoxin Res 2001;7:292-6.
- Kawazoe N, Watabe M, Masuda Y, Nakajo S, Nakaya K. Tiami1 is involved in the regulation of bufalin-induced apoptosis in human leukemia cells. Oncogene 1999;18:2413-21.
- Hill PA, Tumber A, Meikle MC. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology 1997;138:3849-58.
- Mizukami S, Kikuchi K, Higuchi T, Urano Y, Mashima T, Tsuruo T, et al. Imaging of caspase-3 activation in HeLa cells stimulated with etoposide using a novel fluorescent probe. FEBS Lett 1999;453:356-60.
- Adams JM, Cor S. The Bcl-2 protein family: arbiters of cell survival. Science 1998;281:1322-6.
- Hunt A, Evan G. Till death us do part. Science 2001;293:1784-5.
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998;12:2973-83.
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay aging in metazoans. Nature 2004;430:686-9.
- Haim Y. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004;305:390-2.
- Fei XF, Wang BX, Tashiro S, Li TJ, Ma JS, Ikejima T. Apoptotic effects of ginsenoside Rh2 on human malignant melanoma A375-S2 cells. Acta Pharmacol Sin 2002;23:315-22.
- Charrier L, Jarry A, Toquet C, Chedorge M, Denis M, Vallette G, et al. Growth phase-dependent expression of ICAD-L/DFF45 modulates the pattern of apoptosis in human colonic cancer cells. Cancer Res 2002;62:2169-74.
- Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci USA 1986;83:5214-8.
- Wan CH, Wang C, Cheng HY, Yang MS, Fong WF. Triptolide induces Bcl-2 cleavage and mitochondria dependent apoptosis in p53-deficient HL-60 cells. Cancer Lett 2005.1-11.
- Susin SA, Zamzami N, Kroemer G. Mitochondria as regulators of apoptosis: doubt no more. Biochim Biophys Acta 1998;1366:151-65.
- Enari M, Sakahira H, Yokoyama H, Okava K, Aiwamateu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998;391:43-50.
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998;391:96-9.
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier G, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994;371:346-7.
- Muneesh T, Long TQ, Karen OR, Serge D, Zhi Z. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Cell 1995;81:801-9.
- Homayoun V, Scott KD, Elinor NE, Shin-Ichiro I, Roy A. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 2001;107:149-59.
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimeriza-tion with Bax. Nature 1994;369:321-3.
- Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax independent inhibition of apoptosis by Bcl-XL. Nature 1996;379:554-6.
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998;281:1322-6.
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature 1997;387:773-6.
- Yang J, Liu XS, Kapil B, Caryn NK. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997;275:1129-32.
- Kluck RM, Bossy WE, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132-6.
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995;80:293-9.
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683-94.
- Yang YC, Hsu YT, Wu CC, Chen HT, Chang MS. Silencing of astrin induces the p53-dependent apoptosis by suppression of HPV18 E6 expression and sensitizes cells to paclitaxel treatment in HeLa cells. Biochem Biophys Res Commun 2006;343:428-34.
- Zhang QY, Jiang M, Zhao CQ, Yu M, Zhang H, Ding YJ, et al. Apoptosis induced by one new podophyllotoxin glucoside in human carcinoma cells. Toxicology 2005;212:46-53.
- Vercau LD, Beyaert R, Denecker G, Goossens V, Van LG, Declercq W. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998;187:1477-85.