
Cysteinyl leukotriene receptor 1 is involved in N-methyl-D-aspartate-mediated neuronal injury in mice1
Introduction
Excitotoxicity, one important determinant in various diseases of the central nervous system (CNS), is involved in acute ischemic brain injury[1–3] and can initiate postischemic inflammation by inducing the expression of pro-inflammatory molecules/mediators[4–6]. Mediators in postischemic inflammation include 5-lipoxygenase (5-LOX) metabolites eg, cysteinyl leukotrienes (CysLT, including LTC4, LTD4 and LTE4)[7–11]. We have recently indicated that in cultured rat primary neurons, in vitro ischemic-like injury induces endo-genous excitatory amino acid (glutamate) release; the released glutamate activates 5-LOX via the N-methyl-D-aspartate (NMDA) receptor to produce CysLT, which then induce neuron responses[12]. These findings show one aspect of the interaction between 5-LOX/CysLT and excitotoxicity. However, as another aspect, whether the 5-LOX metabolites, CysLT, modulate excitotoxicity is still not clear.
The actions of CysLT are mediated by stimulating their receptors. The cloned CysLT receptors consist of 2 subtypes: CysLT1 and CysLT2 receptors; both of them G-protein coupled receptors[13]. The human CysLT1 receptor is localized in the airway of smooth muscle cells, lung macrophages, mast cells, eosinophils and mononuclear cells as detected by in situ hybridization or immunohistochemistry[14,15]. We have recently reported that CysLT1 receptor is primarily expressed in cerebral microvascular endothelial cells, and the expression is induced in the neuron- and glial-appearing cells after traumatic injury[16]. Moreover, CysLT1 receptor antagonists, pranlukast (ONO-1078) and montelukast, possess neuroprotective effects on focal cerebral ischemia in rats and mice[17–20]. These findings indicate the involvement of the CysLT1 receptor in brain injury. In addition, we found that pranlukast protected against global cerebral ischemia in rats and inhibited the increased expression of the NMDA receptor subunit NR2A, suggesting an interaction between CysLT1 and NMDA receptors[19]. As indirect evidence, pranlukast attenuates brain injury induced by NMDA microinjection into rat cortex, suggesting that the CysLT1 receptor may modulate NMDA-induced neurotoxicity[21]. However, since the CysLT1 receptor is not expressed in the neurons in a normal brain[16], this modulation should be proven by direct evidence.
To determine whether and how the CysLT1 receptor is involved in excitotoxicity, we induced brain injury by NMDA (one of the exogenous excitatory amino acids) microinjection in the cortex. Then we observed CysLT1 receptor expression and the effect of pranlukast, a selective antagonist of CysLT1 receptor, in mice. An NMDA receptor antagonist, ketamine, and an antioxidant with neuroprotective effect for cerebral ischemia, edaravone (MCI-186, 3-methyl-1-phenyl-2-pyrazolin-5-one)[22,23], were used as controls.
Materials and methods
Materials Pranlukast (ONO-1078) was kindly provided by Dr Masami TSUBOSHIMA (Ono Pharmaceutical Co, Osaka, Japan); NMDA and 2,3,5-triphenylterazolium chloride (TTC) were purchased from Sigma (St Louis, MO, USA); edaravone was obtained from Hangzhou Conba Pharmaceutical Co (Hangzhou, China); ketamine was purchased from Shanghai Bio-Chem Co (Shanghai, China); Trizol for extracting RNA was from Bio Basic Inc (Mississauga, Ontario, Canada); chemicals for RT-PCR were from Takara Co (Kyoto, Japan); polyclonal rabbit anti-human CysLT1 antibody was purchased from Cayman Chemicals (Ann Arbor, MI, USA); mouse monoclonal antibodies against neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) and FITC-conjugated goat anti-rabbit IgG and Cy3-conjugated goat anti-mouse IgG were from Chemicon (Temecula, California, USA); cultured human umbilical vein endothelial cells (EA.hy926 cells) were provided by Dr Cora-Jean S EDGELL (University of North Carolina, USA) and human neuroblastoma SK-N-SH cells were purchased from the Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China); biotinylated goat anti-rabbit IgG was purchased from Zhongshan Biotech Co (Beijing, China). Other reagents were commercial products with analytic purity.
NMDA microinjection Male Kunming mice weighing 25–30 g were purchased from the Shanghai Experimental Animal Center (Shanghai, China; Certificate N
The mice were anesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg) and immobilized on a stereotaxic frame (SR-5, Narishige, Tokyo, Japan). The dura overlying the parietal cortex was exposed, and NMDA [50–150 nmol in 0.5 µLof sterile 0.1 mol/L phosphate buffered solution (PBS), pH 7.4] or PBS (0.5 µL) alone was injected with a microinjector into the parietal cortex at a site 1.5 mm caudal to the bregma, 4.0 mm from the midline and 0.8 mm below the dural surface[24]. Injections were made over a period of 8 min, and the microinjector was left in place for an additional 10 min to minimize the back-flux of NMDA and then removed. The rectal temperature was maintained at 37.0±0.5 °C with a heating pad and a heating lamp during the surgical procedure. After the surgery, the mice were kept in a recovery box with heating lamps to maintain body temperature and then returned to their cages.
To observe the effects of the agents, the mice were intraperitoneally injected with pranlukast (0.01 and 0.1 mg/kg), ketamine (30 mg/kg), edaravone (9 mg/kg), and saline (control) at 30 min before and 30 min after NMDA injection.
Histopathological examination The mice were anesthetized with chloral hydrate and decapitated 24 h after NMDA or PBS injection. The brains were quickly removed and cut into 1 mm-thick coronal slices. The slices were stained with 0.5% 2,3,5-triphenylterazolium chloride (TTC) at 37 °C for 30 min in the dark and then fixed by 10% buffered formalin. The stained slices with the caudal facing upwards were photographed with a digital camera (Panasonic CP 230, Matsushita, Fukuoka, Japan) and recorded in a computer. The regions completely lacking TTC-staining were defined as tissue lesions. The lesion and hemisphere area of each slice were quantified using an image analysis program (AanlyPower 1.0, Zhejiang University, Hangzhou, China). The total lesion volume for each brain was calculated by summation of the corrected lesion volumes [lesion area×thickness (1 mm)] of all slices as described by Lin et al[25]. Hemispheric swelling representing brain edema was indirectly determined as the percentage increase of the lesioned hemisphere volume.
In another series, the mice were anesthetized with chloral hydrate and then perfused transcardially with 4% paraformaldehyde after pre-washing with saline. The brains were removed, post-fixed in the same fixative and embedded in paraffin; 5 µm or 8 µm-thick coronal sections were cut by cryomicrotomy (CM1900, Leica, Wetzlar, Germany). Then the 5 µm-thick sections were stained with hematoxylin and eosin (H&E) for light microscopic examination. The densities of the neurons in the cortex and hippocampal CA1 regions (1.8–2.0 mm caudal from bregma) were counted using the image analysis program described above.
RT-PCR The brain tissues (from the region 0.5–2.5 mm caudal from bregma and the corresponding region of the contralateral hemisphere) were dissected on ice and the total RNA was extracted from the tissue samples using Trizol reagents according to the manufacture’s protocol. RNA purity and yield were determined by UV spectrophotometry (Bio-Rad Smart Spec 3000, Hercules, CA, USA). For cDNA synthesis, aliquots of total RNA (2 µg) were mixed with 0.2 µg random hexamer primer, 20 U RNasin, 1 mmol/L dNTP and 200 U M-MuLV reverse transcriptase in 20 µL of the reverse reaction buffer. The mixture was incubated at 42 °C for 60 min and then at 72 °C for 10 min to inactivate the reverse transcriptase. As a negative control, the reaction was performed with the absence of the reverse transcriptase.
The RT-cDNA temple (1 µL) underwent PCR (Bio-Rad, USA) in a 20 µL reaction mixture containing 1×PCR buffer, 200 µmol/L dNTP, 1.5 mmol/L MgCl2, 20 pmol of each primer and 0.5 U of Taq DNA polymerase. Cycling parameters were as follows: 94 °C for 2 min, followed by 33 cycles of 94 °C for 30 s, 63 °C for 30 s and 72 °C for 30 s, with a final extension step of 72 °C for 10 min. The primer pairs for the mouse CysLT1 receptor were derived from the published cDNA sequence[26] and synthesized by Sangon Biotech Co (Shanghai, China): 5'-CAA CGA ACT ATC CAC CTT CACC-3' as sense and 5'-AGC CTT CTC CTA AAG TTT CCAC-3' as antisense. The primers for β-actin were 5'-GTC GTA CCA CAG GCA TTG TGA TGG-3' as sense and 5'-GCA ATG CCT GGG TAC ATG GTG-3' as antisense. The product sizes were 164 bp and 490 bp, respectively. The PCR products in 10 µL were separated by electrophoresis on a 2% agarose gel containing ethidium bromide and photographed. The optical density of the bands was determined by an image analysis system (Bio-Rad, USA). The amount of CysLT1 receptor mRNA was calculated as the ratio over β-actin.
CysLT1 receptor specific immunohistochemical analysis To confirm the specificity of the polyclonal rabbit anti-human CysLT1 receptor antibody used in the immunohistochemical staining of mouse brain tissues, we collected protein samples from normal mouse brains as well as from the cultured human umbilical vein endothelial cells (EA.hy926 cells) and human neuroblastoma SK-N-SH cells for Western blotting analysis. The mouse brain samples were homogenized and cell samples were sonicated in lysis buffer. The lysates were then centrifuged at 15 000×g at 4 °C for 30 min and the supernatant was used. Protein samples (50 μg) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to the nitrocellulose membranes. Then, the membranes were blocked by 5% bovine serum albumin (BSA) and reacted with a rabbit polyclonal antibody against the CysLT1 receptor (1:2000) and peroxidase-conjugated goat anti-rabbit IgG (1:2000) after repeated washing. Finally, the protein bands were visualized by enhanced chemiluminescence.
Immunohistochemical detection of the CysLT1 receptor was performed on 8 µm-thick coronal sections (1.8–2.0 mm caudal from bregma). After 3 washes with PBS, the sections were incubated with 0.3% hydrogen peroxide in methanol at room temperature for 30 min to block the reactivity of endogenous peroxidase. The sections were washed several times in the PBS, pre-incubated with 5% normal goat serum for 2 h to reduce non-specific staining, and then reacted with the polyclonal rabbit anti-human CysLT1 receptor antibody (1:150) overnight at 4 oC. Control sections were treated with normal goat serum instead of the primary antibody. After repeated washing in PBS, the sections were reacted for 1 h with the secondary antibody (1:200), biotinylated goat anti-rabbit IgG, followed by reaction for 1 h with avidin-biotin-horseradish peroxidase complex (1:200). Finally, the sections were exposed for 2–5 min to 0.05% 3,3'-diaminobenzidine and 0.03% hydrogen peroxide and examined by a light microscope (Olympus BX51, Olympus, Tokyo, Japan).
To visualize the localization of the CysLT1 receptor in different cell types, double immunofluorescence was employed. Briefly, non-specific binding of IgG was blocked with 5% normal goat serum for 2 h at room temperature; each section was incubated overnight at 4 °C with a mixture of rabbit polyclonal antibody against the CysLT1 receptor and mouse monoclonal antibodies against NeuN (a specific marker of neurons) or GFAP (a specific marker of astrocytes). Then, the sections were incubated with the mixture of FITC-conjugated goat anti-rabbit IgG and Cy3-conjugated goat anti-mouse IgG and observed under a fluorescence microscope (Olympus BX51, Japan).
Statistical analysis All values are presented as mean±SD. One-way ANOVA (Student-Newman-Keuls) was performed for statistical analysis using the SPSS software package (version 10.0 for Windows; SPSS, Chicago, Illinois, USA). P<0.05 was considered statistically significant.
Results
Effect of pranlukast on NMDA-induced brain injury To confirm the involvement of the CysLT1 receptor in excitotoxicity, we observed the effect of its antagonist pranlukast, on NMDA-induced brain injury in comparison with ketamine and edaravone. NMDA 50, 100, and 150 nmol dose-dependently increased lesion volume (TTC staining) and the lesioned hemisphere volume (indicating brain edema) 24 h after microinjection (P<0.01, Figure 1A–1C). Pranlukast 0.1 mg/kg, edaravone 9 mg/kg and ketamine 30 mg/kg significantly attenuated NMDA-induced (150 nmol) injury (P<0.05 or 0.01, Figure 1D–1F). The histopathological examination showed that NMDA microinjection induced serious pyknotic nuclei and deeply stained cells in the ipsilateral cortex and hippocampal CA1 [not CA3 and dentate gyrus (DG)] region as detected by H&E staining (Figure 2A,2B), and significantly reduced the density of neurons in the cortex (50–150 nmol; Figure 2C) and hippocampal CA1 region (100 and 150 nmol; Figure 2D). Pranlukast (0.1 mg/kg), edaravone (9 mg/kg) and ketamine (30 mg/kg) significantly attenuated NMDA (150 nmol)-reduced density in the cortex (Figure 2E) or hippocampal CA1 region (Figure 2F). These results indicates that the protective effect of pranlukast at 0.1 mg/kg on NMDA-induced brain injury is similar to ketamine and edaravone.


CysLT1 receptor mRNA expression after NMDA microinjection NMDA (100 and 150 nmol) significantly increased the expression of the CysLT1 receptor mRNA, in the injured region of the mouse brain 24 h after NMDA microinjection (P<0.05 or 0.01; Figure 3A, 3D), but NMDA at 50 nmol did not significantly affect the expression (data not shown). Pranlukast (0.1 mg/kg) and ketamine (30 mg/kg) significantly inhibited NMDA (150 nmol)-increased expression of the CysLT1 receptor mRNA (P<0.05) but edaravone did not show this effect (P>0.05; Figure 3F). Otherwise, in the corresponding contralateral region, the expression of the CysLT1 receptor mRNA was not changed (Figure 3E). This result indicated that NMDA increased the expression of the CysLT1 receptor mRNA, which was inhibited by the NMDA receptor antagonist ketamine and the CysLT1 receptor antagonist, pranlukast, but not by the antioxidant edaravone.

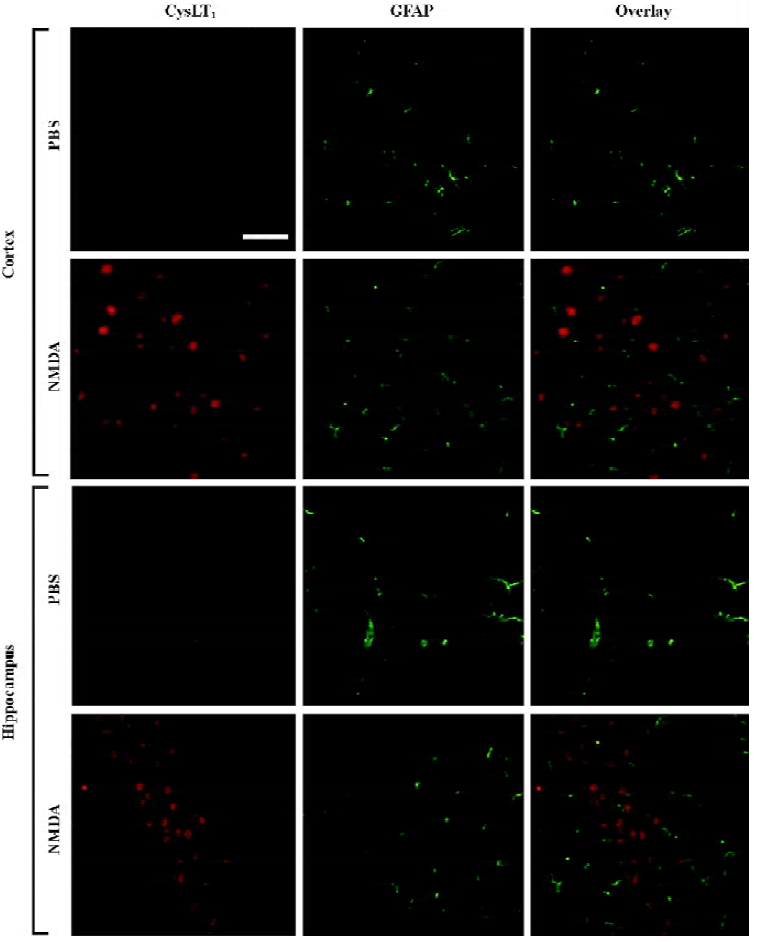
Distribution of the CysLT1 receptor immunopositive cells after NMDA microinjection Western blotting analysis confirmed the specificity of a polyclonal rabbit anti-human CysLT1 receptor antibody used in the mouse brain because the same bands were found in the mouse brain samples and the samples from cultured human umbilical vein endothelial cells (EA.hy926 cells) or human neuroblastoma SK-N-SH cells (Figure 4E). The band was closed to 43 kDa, which was consistent with previously published results using the same polyclonal antibody[27,28]. Using the antibody, we detected the distribution of CysLT1 receptor protein 24 h after NMDA (150 nmol) microinjection by immunohistochemistry. The result showed that CysLT1-positive cells were significantly increased in the cortex and hippocampal CA1 region after NMDA excitotoxic damage (Figure 4A–4D), but not in the hippocampal CA3 or DG region. Pranlukast (0.1 mg/kg) and ketamine (30 mg/kg) reduced CysLT1-positive cells, but edaravone (9 mg/kg) did not show this effect (Figure 4A–4D). To determine whether the increased expression of the CysLT1 receptor is distributed in neurons or astrocytes, we performed double immunofluorescence. The result showed that CysLT1 receptor immunoreactivity was mainly localized in NeuN-positive neurons in the NMDA (150 nmol)-injected cortex and hippocampal CA1 region (Figure 5). However, no apparent change was found in GFAP-positive astrocytes; the CysLT1 receptor was much less expressed in the astrocytes (Figure 6).


Discussion
The most important finding in the present study is that the CysLT1 receptor is involved in brain excitotoxicity. This involvement is evidenced by the upregulation of the CysLT1 receptor after NMDA microinjection and the attenuation of NMDA insult by a CysLT1 receptor antagonist, pranlukast. Therefore, our study shows a possible interaction between the excitotoxicity and the inflammation related to CysLT in the brain.
Our immunohistochemical results indicate that CysLT1 receptor expression is induced by NMDA microinjection and mainly localized in the neurons, but not in the astrocytes in the injured regions. This finding is consistent with those of our recent studies. We found that the CysLT1 receptor was primarily distributed in microvascular endothelial cells in the human brain, and an inducible expression was detected in the neuron- and glial-appearing cells in the brain specimens from patients with brain trauma or tumors[16]. In rats and mice with focal cerebral ischemia, CysLT1 receptor expression was largely increased in the ischemic core 24 h after ischemia, and the increased expression was mainly localized in NeuN-positive neurons and much less in GFAP-positive astrocytes (unpublished data). These findings suggest that the CysLT1 receptor may mediate various brain injuries, such as trauma, ischemia and tumors, as well as chemically-induced excitotoxicity in the present study.
Interestingly, the increased CysLT1 receptor expression induced by NMDA is not only inhibited by the NMDA receptor antagonist, ketamine, but also by the CysLT1 receptor antagonist, pranlukast. Since none of the agents affect the expression of a CysLT1 receptor mRNA, in the contralateral brain region from the NMDA-treated mice, it can be excluded that ketamine or pranlukast directly inhibits the expression. Inhibition by ketamine reasonably results from the blockage of NMDA actions. However, why pranlukast also inhibits CysLT1 receptor expression is unclear. One possible explanation might be that attenuation of brain injury by pranlukast may secondarily reduce CysLT1 receptor expression; however, this explanation is not supported by the effect of edaravone that attenuated NMDA-induced injury, but did not inhibit the expression. Another explanation might be that this phenomenon may be a special effect of CysLT1 receptor antagonists. We have found that montelukast, another CysLT1 receptor antagonist, also inhibited CysLT1, but not CysLT2 receptor mRNA expression in the lungs with eosinophilic inflammation from asthmatic mice[29]. Because interleukin-5 (IL-5) upregulates CysLT1 receptor expression[30] and pranlukast inhibits IL-5 produc-tion[29,31], inhibition of CysLT1 receptor expression by pranlukast might result from its effect on upregulation by IL-5.
Among the agents used in the present study, ketamine is applied to confirm NMDA receptor activation, pranlukast is to confirm CysLT1 receptor activation, and edaravone is to distinguish the differences from pranlukast. Ketamine is a potent non-competitive NMDA receptor antagonist that has been shown to protect neurons from excitotoxic injury after cerebral ischemia[32,33], trauma[34] or injection of excitotoxins[35,36]. In the present study, the inhibition of all the responses to NMDA by ketamine confirmed that NMDA-induced responses are mediated by NMDA receptor activation, similar to the reported results[36,37]. For the effect of pranlukast, we used 2 doses of pranlukast; 0.01 mg/kg was a nearly ineffective dose and 0.1 mg/kg was the most effective dose in the experiments of cerebral ischemia[17,18,20]. The results showed dose-dependency; only 0.1 mg/kg exerts effect on brain injury. In addition, edaravone (MCI-186, 3-methyl-1-phenyl-2-pyrazolin-5-one) is a clinically available neuropro-tective agent for the treatment of stroke with activity reducing free radicals[22,23,38,39]. We found that edaravone had a different effect from pranlukast; it attenuated NMDA-induced brain injury, similar to its neuroprotective effect on cerebral ischemia[23,38,39], but did not affect CysLT1 receptor expression. Therefore, this difference supports that the effect of pranlukast on CysLT1 receptor expression might be special.
Excitotoxicity is a common injurious factor involved in many CNS diseases including cerebral ischemia. The present study indicates one aspect of the interaction between excitotoxicity and 5-LOX/CysLT pathway: the CysLT1 receptor is upregulated and plays a role in excitotoxicity. As another aspect, we recently found that 5-LOX was upregulated by excitotoxicity[12]. Taken together, excitotoxicity initiates post-injury inflammation by enhancing both pro-inflammatory molecules (like 5-LOX) and inflammatory responses (like strengthening the action of CysLT).
Acknowledgments
We thank Dr Masami TSUBOSHIMA (Ono Pharmaceutical Co Ltd, Osaka, Japan) for providing us with pranlukast, Dr Cora-Jean S EDGELL(Pathology Department, University of North Carolina) for providing EA.hy926 cells, and Professor Jian-hong LUO, Department of Neurobiology, School of Medicine, Zhejiang University, for critically reading and commenting on this manuscript.
References
- Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 1999;163:3963-8.
- Gucuyener K, Atalay Y, Aral YZ, Hasanoglu A, Turkyilmaz C, Biberoglu G. Excitatory amino acids and taurine levels in cerebrospinal fluid of hypoxic ischemic encephalopathy in newborn. Clin Neurol Neurosurg 1999;101:171-4.
- Favalli L, Rozza A, Frattini P, Masoero E, Scelsi R, Pascale A, et al. Ischemia-induced glutamate release in rat frontoparietal cortex after chronic alcohol and withdrawal. Neurosci Lett 2002;326:183-6.
- Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience 2000;101:737-44.
- Jander S, Schroeter M, Stoll G. Role of NMDA receptor signaling in the regulation of inflammatory gene expression after focal brain ischemia. J Neuroimmunol 2000;109:181-7.
- Acarin L, Gonzalez B, Castellano B. Decrease of proinflamma-tory molecules correlates with neuroprotective effect of the fluorinated salicylate triflusal after postnatal excitotoxic damage. Stroke 2002;33:2499-505.
- Mabe H, Nagai H, Suzuka T. Role of brain tissue leukotriene in brain oedema following cerebral ischaemia: effect of a 5-lipoxygenase inhibitor, AA-861. Neurol Res 1990;12:165-8.
- Baba T, Black KL, Ikezaki K, Chen KN, Becker DP. Intracarotid infusion of leukotriene C4 selectively increases blood-brain barrier permeability after focal ischemia in rats. J Cereb Blood Flow Metab 1991;11:638-43.
- Rao AM, Hatcher JF, Kindy MS, Dempsey RJ. Arachidonic acid and leukotriene C4: role in transient cerebral ischemia of gerbils. Neurochem Res 1999;24:1225-32.
- Ciceri P, Rabuffetti M, Monopoli A, Nicosia S. Production of leukotrienes in a model of focal cerebral ischaemia in the rat. Br J Pharmacol 2001;133:1323-9.
- Di Gennaro A, Carnini C, Buccellati C, Ballerio R, Zarini S, Fumagalli F, et al. Cysteinyl-leukotrienes receptor activation in brain inflammatory reactions and cerebral edema formation: a role for transcellular biosynthesis of cysteinyl-leukotrienes. FASEB J 2004;18:842-4.
- Ge QF, Wei EQ, Zhang WP, Hu X, Huang XJ, Zhang L, et al. Activation of 5-lipoxygenase after oxygen-glucose deprivation is partly mediated via NMDA receptor in rat cortical neurons. J Neurochem 2006;97:992-1004.
- Brink C, Dahlen SE, Drazen J, Evans JF, Hay DW, Nicosia S, et al. International Union of Pharmacology XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol Rev 2003;55:195-227.
- Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 1999;399:789-93.
- Sarau HM, Ames RS, Chambers J, Ellis C, Elshourbagy N, Foley JJ, et al. Identification, molecular cloning, expression, and characterization of a cysteinyl leukotriene receptor. Mol Pharmacol 1999;56:657-63.
- Zhang WP, Hu H, Zhang L, Ding W, Yao HT, Chen KD, et al. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci Lett 2004;363:247-51.
- Zeng LH, Zhang WP, Wang RD, Wang PL, Wei EQ. Protective effect of ONO-1078, a leukotriene antagonist, on focal cerebral ischemia in mice. Yao Xue Xue Bao 2001;36:148-50. Chinese..
- Zhang WP, Wei EQ, Mei RH, Zhu CY, Zhao MH. Neuropro-tective effect of ONO-1078, a leukotriene receptor antagonist, on focal cerebral ischemia in rats. Acta Pharmacol Sin 2002;23:871-7.
- Zhang LH, Wei EQ. Neuroprotective effect of ONO-1078, a leukotriene receptor antagonist, on transient global cerebral ischemia in rats. Acta Pharmacol Sin 2003;24:1241-7.
- Yu GL, Wei EQ, Zhang SH, Xu HM, Chu LS, Zhang WP, et al. Montelukast, a cysteinyl leukotriene receptor-1 antagonist, dose- and time-dependently protects against focal cerebral ischemia in mice. Pharmacology 2005;73:31-40.
- Zhang LH, Wei EQ. ONO-1078 reduces NMDA-induced brain injury and vascular cell adhesion molecule-1 expression in rats. Acta Pharmacol Sin 2005;26:435-40.
- Toyoda K, Fujii K, Kamouchi M, Nakane H, Arihiro S, Okada Y, et al. Free radical scavenger, edaravone, in stroke with internal carotid artery occlusion. J Neurol Sci 2004;221:11-7.
- Yasuoka N, Nakajima W, Ishida A, Takada G. Neuroprotection of edaravone on hypoxic-ischemic brain injury in neonatal rats. Dev Brain Res 2004;151:129-39.
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA 2001;98:1294-9.
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 1993;24:117-21.
- Ogasawara H, Ishii S, Yokomizo T, Kakinuma T, Komine M, Tamaki K, Characterization of mouse cysteinyl leukotriene receptors mCysLT1 and mCysLT2: differential pharmacological properties and tissue distribution. J Biol Chem 2002; 277: 18 763–8.
- Mazzetti L, Franchi-Micheli S, Nistri S, Quattrone S, Simone R, Ciuffi M, et al. The ACh-induced contraction in rat aortas is mediated by the Cys Lt1 receptor via intracellular calcium mobilization in smooth muscle cells. Br J Pharmacol 2003;138:707-15.
- Ohd JF, Nielsen CK, Campbell J, Landberg G, Lofberg H, Sjolander A. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology 2003;124:57-70.
- Zhang YJ, Zhang L, Wang SB, Shen HH, Wei EQ. Montelukast modulates lung CysLT1 receptor expression and eosinophilic inflammation in asthmatic mice. Acta Pharmacol Sin 2004;25:1341-6.
- Thivierge M, Doty M, Johnson J, Stankova J, Rola-Pleszczynski M. IL-5 up-regulates cysteinyl leukotriene 1 receptor expression in HL-60 cells differentiated into eosinophils. J Immunol 2000;165:5221-6.
- Fukushima C, Matsuse H, Hishikawa Y, Kondo Y, Machida I, Saeki S, et al. Pranlukast, a leukotriene receptor antagonist, inhibits interleukin-5 production via a mechanism distinct from leukotriene receptor antagonism. Int Arch Allergy Immunol 2005;136:165-72.
- Church J, Zeman S, Lodge D. The neuroprotective action of ketamine and MK-801 after transient cerebral ischemia in rats. Anesthesiology 1988;69:702-9.
- Proescholdt M, Heimann A, Kempski O. Neuroprotection of S(+) ketamine isomer in global forebrain ischemia. Brain Res 2001;904:245-51.
- Shapira Y, Lam AM, Eng CC, Laohaprasit V, Michel M. Therapeutic time window and dose response of the beneficial effects of ketamine in experimental head injury. Stroke 1994;25:1637-43.
- Leander JD, Lawson RR, Ornstein PL, Zimmerman DM. N-methyl-D-aspartic acid-induced lethality in mice: selective antagonism by phencyclidine-like drugs. Brain Res 1988;448:115-20.
- Lees GJ. Influence of ketamine on the neuronal death caused by NMDA in the rat hippocampus. Neuropharmacology 1995;34:411-7.
- Lees GJ. Effects of ketamine on the in vivo toxicity of quinolinate and N-methyl-D-aspartate in the rat hippocampus. Neurosci Lett 1987;78:180-6.
- Wu TW, Zeng LH, Wu J, Fung KP. MCI-186: further histochemical and biochemical evidence of neuroprotection. Life Sci 2000;67:2387-92.
- Ikeda T, Xia YX, Kaneko M, Sameshima H, Ikenoue T. Effect of the free radical scavenger, 3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186), on hypoxia-ischemia-induced brain injury in neonatal rats. Neurosci Lett 2002;329:33-6.