
Effects of endothelin ETA receptor blocker LU 135252 on cardiac remodeling and survival in a hypertensive rat model of chronic heart failure1
Introduction
Endothelin-1 (ET-1) is a vasoactive peptide that is produced by the vascular endothelium. ET-1 through 2 principal endothelin receptors, ETA and ETB, exerts a pivotal role in cardiovascular regulation[1]. The ETA receptor is believed to induce proliferation of vascular smooth muscles and myocardial hypertrophy[2], whereas the activation of the endothelial ETB receptor causes vasodilation via a release of nitric oxide and/or prostacyclin from the endothelial cells, and results in antithrombotic and antiproliferative effects[3]. Further, pulmonary ETB receptors are important for the clearance of ET-1[4].
The plasma level of ET-1 is raised in patients and animals with chronic heart failure (CHF), particularly in more advanced CHF[5]. The increase of plasma ET-1 is correlated with the extent of hemodynamic impairment and is a strong independent predictor of mortality[6]. Like angiotensin-converting-enzyme inhibitors and β blockers, in experimental CHF, endothelin blockade prevents left ventricular (LV) remodeling and prolongs survival in rabbits with pacing-induced CHF[7] and in rats with myocardial infarction (MI)-induced CHF[8,9]. However, the clinical trials from the Endothelin Antagonism Bosentan for Lowering Cardiac Events in Heart Failure (ENABLE) study[10] and from the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH) trial[11], both non-selective and selective endothelin-receptor antagonists did not improve cardiac remodeling and clinical symptoms or reduce morbidity and mortality in patients with CHF.
Why did the good news in the experimental study become bad news after clinically effective therapy? Normotensive animal models, which were used in most experimental protocols investigating the effects of endothelin blockade on CHF, might not fully represent the gradually deteriorated and complex clinical situation of heart failure. This could be one explanation for the contrary data from experimental and clinical use. Ischemic and hypertensive heart diseases are commonly associated pathophysiological parameters of heart failure in humans. Stroke-prone spontaneously hypertensive (SHR-SP) rats, a genetic model of hypertension, combined these 2 parameters. This study therefore was performed to determine whether the ETA receptor antagonist LU 135252 prevents cardiac remodeling and improves survival in SHR-SP model of MI-induced heart failure.
Materials and methods
Animals and model of CHF Male SHR-SP rats weighing 280–300 g were used for the experiments. The animals were housed in individual cages under constant humidity and temperature, and exposed to a 12 h light/dark cycle with free access to a standard chow (Ssniff Spezialdiäten GmbH, Soest, Germany) and drinking water. All experiments were approved by the German governmental office dealing with animal protection and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication N
The present study employed a model of MI-induced CHF. MI was induced by permanent ligation of the left descending coronary artery as previously described[12]. The mortality rate was about 40% within the first 24 h. Of the 250 rats that had undergone left coronary artery ligation, 98 died. Sham-operated controls (n=12) underwent the same surgical procedure except that the suture around the coronary artery was not tied.
Dose determination study In order to determine the doses of LU 135252, under chloral hydrate (400 mg/kg, ip) anaesthesia, polypropylene tubes were inserted into the right femoral artery and vein and exteriorized at the nape of the neck in 6 rats. Forty eight hours later, the basal mean arterial blood pressure (MAP) was recorded for 30 min, then an intravenous administration of ET-1, 250 nmol, sufficient to increase MAP by 20 mmHg or more was applied. After this initial pressor test, the other 24 SHR-SP rats were randomly assigned to 1 of 4 groups to receive oral LU 135252 10, 30, and 50 mg·kg-1·d-1 or no treatment (controls) for 4 weeks. At the end of treatment, the MAP was recorded and the pressor responses to exogenous ET-1 were then repeated. The results showed that the pressor response was suppressed and the MAP was 165 mmHg in LU 135252 30 mg· kg-1 ·d-1 and 185 mmHg in the control group, respectively. This degree of suppression of the pressor response was considered a surrogate to sufficient systematical blockade of the ETA receptor. On the basis of these data, the doses of LU 135252, 30 mg· kg-1·d-1, were chosen for the study.
Experimental protocol The surviving MI rats (152) were divided randomly into the untreated group (n=80) and the ETA receptor antagonist LU 135252 treated group (30 mg· kg-1·d-1, n=72). Untreated sham-operated rats (n=12) were used as controls. LU 135252 {[+]-5-2-[4, 6-dimethoxy-pyrimidin-2-yloxy]-3-methoxy-3,3-diphenyl-propionic acid} was provided by Knoll AG (Ludwigshafen, Germany) and was dissolved in the drinking water according to the manufacturer’s instructions; treatment was started 24 h after MI and continued for up to 6 weeks post MI. At the end of the treatment period, arterial, venous and LV catheters were chronically implanted. Twenty-four hours later, hemodynamic signals were recorded in the conscious rats. At the end of the recording, the rats were sacrificed and the hearts were taken out for morphological examinations.
Hemodynamic studies Under chloral hydrate (200 mg/kg, ip) anaesthesia, polypropylene tubes (Portex, London, UK) were inserted into the right femoral artery and vein and a specially constructed pig-tail catheter consisting of PP 10 and PP 50 polypropylene tubes via the right common carotid artery were introduced in the left ventricle. The catheters then were exteriorized and anchored at the posterior neck region.
Blood pressure, heart rate (HR) and LV pressure were recorded 24 h after catheterization in the conscious rats. The arterial and LV catheters were connected to pressure transducers (DTX/Plus, Spectramed Inc, Oxnard, CA, USA), and all hemodynamic signals were processed by 2 pressure processors (Gould Inc, Valley View, OH, USA). The output signals (MAP, HR and LV pressure) from these pressure processors were recorded on a pen recorder (Gould Series 2000, Gould Inc) and analysed by a computer-based recording and analyzing system (MEGA). The rats were allowed to get acquainted to the recording circumstances for 30 min before hemodynamic measurements.
Left ventricular end-diastolic pressure (LVEDP) and the maximum rate of rise of LV pressure (LV dp/dtmax) were calculated offline from the LV pressure signal by the MEGA program. MAP, HR, LVEDP and LV dp/dtmax were averaged over a 5 min period to be used in the statistical analysis.
Cardiac morphological examinations After the recording of the hemodynamic signals, the heart was arrested in diastole by an intravenous injection of 1% KCl solution under ether anaesthesia. The heart was excised and the atria and large vessels removed. The heart was cleaned and weighed, placed in 4% phosphate-buffered formalin (0.15 mol/L NaCl) for at least 24 h and then cut transversely into 5 sections of approximately identical thickness from the apex to the base. These sections were transferred into 10% phosphate-buffered formalin and were kept overnight. After dehydration, the sections were embedded in paraffin and cut in serial 7-µm-thick slices. The slices were mounted onto glass slides and were then stained with picrosirius red.
Measured morphological parameters were total heart weight (HW), infarct size, septal thickness, left ventricular circumference (LVC), left ventricular inner diameter (LVD) and interstitial collagen content (ICC). For these measure-ments, a computerised morphometric system (Quantimet 570, Leica, Cambridge, UK) was used.
Survival After the initiation of LU 135252 treatment, the cages were inspected daily for dead rats in order to calculate the survival rate. The hearts of the dead animals were removed and fixed in formalin solution for subsequent determination of infarct size.
Statistical analysis All data are expressed as mean±SEM. Comparisons among sham-operated, MI untreated and MI LU 135252-treated groups were performed by one-way analysis of variance (ANOVA) followed by a post hoc Bonferroni test. Mortality was compared between the treated and untreated groups using the Kaplan-Meier analysis of survival followed by the Log-rank test (Cox-Mantel). Values of P<0.05 were considered to be statistically significant.
Results
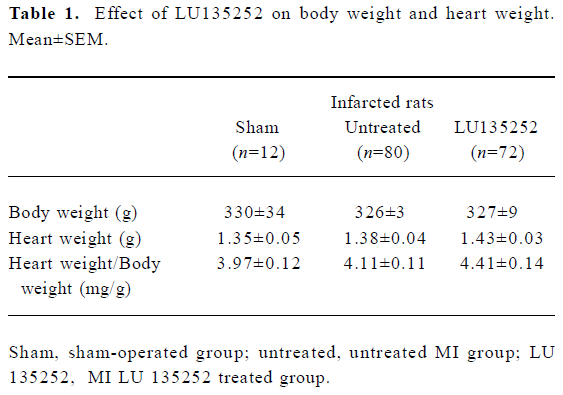
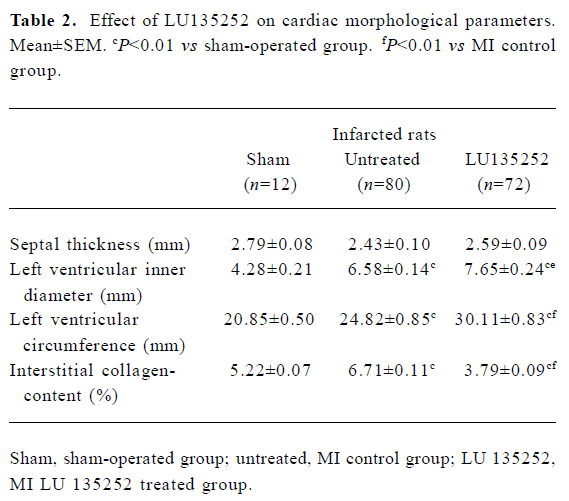
Characteristics of untreated infarct rats Six weeks after induction of MI in the untreated MI SHR-SP group, the HW and HW to body weight ratio (HW/BM) were still slightly higher compared to the age-matched sham-operated SHR-SP, indicating that reactive hypertrophy occurred in remaining myocardium despite 40.7% of the LV myocardium being replaced by scar tissue (Tables 1, 2). In addition, in the untreated MI rats, the ICC of the non-infarcted myocardium significantly increased, the LVD and LVC were enlarged (all P<0.01), while septal thickness slightly decreased, indicating the development of LV dilation in SHR-SP with CHF 6 weeks after MI.
Full table
Full table
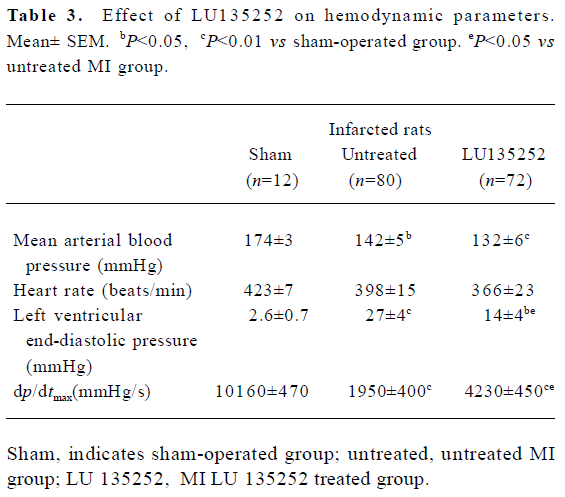
In the untreated MI animals, MAP fell by 32 mmHg, LV dp/dtmax decreased by 80% and LVEDP significantly increased compared to the sham-operated controls (all P< 0.01).
These data indicated that 6 weeks after coronary artery ligation, the untreated animals developed severe CHF, evidenced by an augmented LVEDP and LV cavity dilation, as well as deterioration of LV functions assessed by cardiac hemodynamics (Tables 2, 3).
Full table
Survival Six weeks after MI or sham surgery, the survival of the sham-operated group was 85%, with 10 of 12 rats staying alive. MI markedly decreased the survival; the survival of the untreated MI group was only 11% (n=9/80, sham vs untreated; P<0.01). LU 135252 treatment trended towards no effect on survival with respect to the untreated MI group (n=9/72, 13% vs 11%; P=NS).
Infarct size Infarct size was measured in all animals. None of the sham-operated rats showed evidence of MI by gross inspection; the coronary ligation animals showed clear microscopic evidence of transmural infarction.
In the present study, the size of the infarct area was expressed as a percentage of the total circumference of the left ventricle. Three animals with an infarct size of less than 30% were excluded from the study. The infarct size was 40.7%±3.0% in the untreated MI group and 41.2%±2.8% in the LU 135252-treated group. No significant differences in the infarct size of the 2 groups were detected.
Heart mass Heart mass was evaluated by determining the HW and the HW/BM ratio. Although 40.7% the of LV myocardium was replaced by paper-thin scar tissue, HW and HW/BW were still slightly higher in the untreated infarct group compared to the sham-operated animals, suggesting that reactive hypertrophy had occurred in the remaining myocardium. LU 135252 treatment trended to increase HW and HW/BW compared to the untreated MI group (Table 1).
LV size The LVD and the circumference LVC of the left ventricle served as markers for left LV dilation.
In the untreated MI group, the LVD increased by 60% compared to the sham-operated controls (P<0.01). LU 135252 treatment significantly increased LVD compared to the untreated MI group (P<0.05; Table 2).
The circumference of the LVC increased by 20% in the untreated MI group compared to sham-operated controls (P<0.01). LU 135252 treatment markedly increased LVC compared to the untreated MI group (P<0.01; Table 2).
In the untreated MI group, septal thickness decreased compared to the sham-operated controls. The differences in septal thickness between the LU 135252-treated and untreated MI groups did not reach statistical significance (Table 2).
ICC When compared to the age-matched sham-operated SHR-SP, the untreated MI rats showed a significant increase in ICC (P<0.01). The ICC significantly decreased in the LU 135252 treatment group compared to the untreated MI group (P<0.01; Table 2).
Systemic hemodynamics Six weeks after induction of MI, the MAP was lowered by 32 mmHg in the untreated MI group compared to the sham-operated controls (P<0.01), and was slightly further lowered in the LU 135252 treated group (Table 3).
The MI and LU 135252 treatment tended to reduce HR, but the differences between the groups did not reach statistical significance (Table 3).
Cardiac hemodynamics LVEDP significantly increased in the untreated MI group compared to the sham-operated controls (P<0.01). LU 135252 treatment decreased LVEDP (P<0.05). LV dp/dtmax as a marker of LV myocardial contractility was severely impaired in the untreated MI group compared to the sham-operated controls (P<0.01). LU 135252 therapy improved myocardial contractility compared to the untreated MI group (P<0.05; Table 3).
Discussion
The primary purpose of this study was to elucidate why the endothelin receptor blockade failed to improve the outcome of CHF in the ENABLE study[10] and EARTH trial[11], but gave promising data in the experimental studies. To address this question, we used the SHR-SP rat model of infarct-induced heart failure.
The compound LU 135252 has been investigated as a selective ETA receptor antagonist with good oral bioavail-ability and a long duration of action[13]. The Ki is 1.4 nmol/L for the ETA receptor and 184 nmol/L for the ETB receptor[14]. Thus, LU 135252 has a 130-fold higher affinity to the ETA receptor than to the ETB receptor.
Effects of LU 135252 on cardiac morphology The present study shows that the chronic administration of an endothelin ETA receptor antagonist, LU 135252, did not prevent cardiac hypertrophy, markedly reduced interstitial collagen deposition and caused further dilation of the left ventricle. These results are in agreement with a study by Mulder et al[9] who observed that chronic oral treatment with LU 135252 over 10 weeks beginning 7 d after MI in normotensive rats reduced LV collagen density, but did not prevent cardiac hypertrophy.
Cardiac hypertrophy is associated with 2 remodeling events: an increase in cardiac muscle mass and an abnormal accumulation of fibrillar collagen. The myocardium is composed of cardiac myocytes and non-myocytes including fibroblasts, vascular endothelial cells, vascular smooth muscle cells and macrophages. Soon after birth, cardiac myocytes lose the ability to proliferate, and the heart grows by increasing the size of myocytes and increasing the number of non-myocytes. Although cardiac myocytes make up most of the adult myocardial mass, they comprise only 30% of the total cell number present in the heart; the rest is composed of non-myocytes, and more than 90% of them are fibroblasts. In response to injury or infarction of myocardium, the growth of fibroblasts is stimulated. Fibroblasts express both endothelin ETA and ETB receptor subtypes in nearly equal proportions, and ET-1 increases the synthesis of type I and III collagens[15,16]. In the present study, treatment with the ETA receptor blocker LU 135252 for 6 weeks decreased ICC indicating that the incidence of myocardial fibrosis in SHR-SP may be partly mediated by the activation of the ETA receptor localized on cardiac fibroblasts.
LU 135252 treatment did not diminish cardiac hyper-trophy. This finding is not limited to the selective ETA receptor blockade because Mulder et al[17] reported that the non-selective ET receptor blocker, bosentan, similarly failed to prevent cardiac hypertrophy in MI-induced CHF in Wistar rats. The lack of effect of LU 135252 on cardiac hypertrophy could be due to an incomplete blockade of ETA receptors in cardiac tissues. The dose of LU 135252 used in the present study (30 mg·kg-1·d-1) was chosen after pilot experiments, because more than 50% of pressor responses of ET-1 50 ng/kg iv were suppressed. This degree of suppression of the pressor response is considered a surrogate to sufficient systematical blockade of the ETA receptors. Accordingly, the present dose of 30 mg·kg-1·d-1 was considered to sufficiently antagonize the local paracrine ET system of the myocardium. Furthermore, in the present study LU 135252 reduced blood pressure confirming effective receptor blockade. Thus, inadequate dosing is unlikely to account for the effects of the drug.
In addition, it was observed that LU 135252 treatment significantly increased LVD and LVC compared to the untreated MI rats indicating that the LV was further dilated. The exact mechanism still remains to be further elucidated. However, LU 135252 treatment decreased the ICC. The marked LV dilation in the LU 135252-treated animals could be due to an “over-suppression” of ICC. An appropriate increase of myocardial collagen content after MI might be beneficial to a certain degree since it is an adaptive mechanism of the myocardium to maintain functional elasticity and strength. ET blockade could lead to a sustained suppression of this adaption of the myocardial collagen network. Several studies have demonstrated that the LV dilation after MI is one of the strongest predictors of morbidity (eg progression to heart failure) and mortality[9]. Therefore, the survival of the LU 135252-treated rats was not improved.
As previously mentioned, the ETB receptors are localized on the vascular endothelium and mediate vasorelaxation by increasing the formation of prostacyclin and nitric oxide (NO) as well as the clearance of circulating ET-1. The ETB receptors are present on smooth muscle cells as well where they also mediate vasoconstriction. ETB receptor inhibition reduces ET-1 clearance and NO-mediated vasodilatation. ETB receptor inhibition may abrogate the beneficial effects of the ETA receptor blockade. Therefore, the lack of effect of LU 135252 on cardiac remodeling cannot exclude the role of ETB receptor inhibition. However, Mulder et al[9] reported that LU 135252 used at a dose of 30 mg/kg per day blocked the ETA-mediated vasoconstrictor response without modifying the ETB-mediated dilator response.
Effects of LU 135252 on hemodynamics The results of the present study indicate that treatment with the ETA receptor antagonist LU 135252, leads to an improvement of LV function in SHR-SP with MI-induced CHF. The hemodynamic response to LU 135252 is characterized by an increase of myocardial contractility and a reduction in LVEDP. The present results are in accordance with the results of Sakai et al[8] who reported that the long-term administration of ETA receptor antagonist BQ123, improved LV dp/dtmax and decreased LVEDP. They are also in agreement with those of Spinale et al[7] who demonstrated improvements in myocyte contractile function and LV fractional shortening after chronic treatment with ETA receptor antagonist PD 156707 in a rabbit model of pacing-induced CHF.
ET-1 is a positive inotropic agent on the intact LV and isolated cardiac myocytes from normal animals[18]. This effect is largely mediated by the ETA receptor[19]. Plasma and myocardial ET-1 levels are increased in CHF, and the effect of ET-1 on normal myocardial contraction may be altered in pathological states. Sakai et al[8] reported that after MI in rats, the elevated plasma and myocardial ET-1 levels were associated with a decline in LV myocardial contractility, and the ETA receptor blockade increased LV dp/dtmax and improved myocardial contractility. The present study confirms their findings in the SHR-SP model of MI-induced CHF.
LU 135252 decreases LVEDP and arterial blood pressure, indicating a decrease in cardiac preload and afterload. This, together with the limitation of LV remodelling, results in an increased stroke volume and cardiac output and prevents the deterioration of global LV function.
In conclusion, the endothelin ETA receptor blockade with LU 135252 reduces LVEDP and improves LV dp/dtmax, how-ever, LU 135252 does not affect survival, tends to increase HW, significantly increases LVD and LVC and causes further dilation of the left ventricle, indicating that LU 135252 treatment worsens cardiac remodeling in this novel SHR-SP model of CHF. The present study could provide one explanation for the contrary data from experimental and clinical use of endothelin receptor antagonists.
References
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1998;332:411-5.
- Yang Z, Krasnici N, Lüscher TF. Endothelin-1 potentiates human smooth muscle cell growth to PDGF: effects of ETA and ETB receptor blockade. Circulation 1999;100:5-8.
- Haynes WG, Webb DJ. Endothelium-dependent modulation of responses to endothelin-1 in human veins. Clin Sci 1993;84:427-33.
- Dupuis J, Stewart DJ, Cernacek P, Gosselin G. Human pulmonary circulation is an important site for both clearance and production of endothelin-1. Circulation 1996;94:1578-84.
- McMurray JJ, Ray SG, Abdullah I, Dargie HJ, Morton JJ. Plasma endothelin in congestive heart failure. Circulation 1992;85:1374-9.
- Omland T, Lie RT, Aakvaag A, Aarsland T, Dickstein K. Plasma endothelin determination as a prognostic indicator of 1-year mortality after acute myocardial infarction. Circulation 1994;89:1573-9.
- Spinale FG, Walker JD, Mukherjee R, Iannini JP, Keever AT, Gallagher KP. Concomitant endothelin receptor subtype-A blockade during the progression of pacing-induced congestive heart failure in rabbits. Circulation 1997;95:1918-29.
- Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 1996;384:353-5.
- Mulder P, Richard V, Bouchart F, Derumeaux G, Munter K, Thuillez C. Selective ETA receptor blockade prevent left ventricular remodeling and deterioration of cardiac function in experimental heart failure. Cardiovasc Res 1998;39:600-8.
- Coletta A, Thackray S, Nikitin N, Cleland JG. Clinical trials update: highlights of the scientific sessions of the American College of Cardiology 2002: LIFE, DANAMI 2, MADIT-2, MIRACLE-ICD, OVERTURE, OCTAVE, ENABLE 1&2, CHRISTMAS, AFFIRM, RACE, WIZARD, AZACS, REMATCH, BNP trial and HARDBALL. Eur J Heart Fail 2002;4:381-8.
- Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the endothelin A receptor antagonist trial in heart failure (EARTH): randomised, double blind, placebo-controlled trial. Lancet 2004;364:347-54.
- Xia QG, Chung O, Spitznagel H, Illner S, Janichen G, Rossius B, et al. Significance of timing of angiotensin AT1 receptor blockade in rats with myocardial infarction-induced heart failure. Cardio-vasc Res 2001;49:110-7.
- Riechers H, Albrecht HP, Amber W, Baumann E, Bernard H, Bohm HJ, et al. Discovery and optimization of a novel class of orally active nonpeptidic endothelin-A receptor antagonists. J Med Chem 1996;39:2123-8.
- Jasmin JF, Lucas M, Cernacek P, Dupuis J. Effectiveness of a nonselective ETA/B and a selective ETA antagonist in rats with monocrotaline-induced pulmonary hypertension. Circulation 2001;103:313-8.
- Booz GW, Baker KM. Molecular singling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res 1995;30:537-43.
- Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardio-vasc Res 1993;27:2130-4.
- Mulder P, Richard V, Derumeaux G, Hogie M, Henry JP, Lallemand F, et al. Role of endothelin in chronic heart failure. Effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation 1997;96:1976-82.
- Mebezaa A, Mayoux E, Maeda K. Paracrine effects of endocardial endothelial cells on myocyte contraction mediated via endothelin. Am J Physiol 1993;265:H1841-6.
- Kasai H, Takanashi M, Takasaki C, Endoh M. Pharmacological properties of endothelin receptor subtypes mediating positive inotropic effects in rabbit heart. Am J Physiol 1994;266:H2220-8.