
Identification of a novel splice variant of human PD-L1 mRNA encoding an isoform-lacking Igv-like domain1
Introduction
The signals provided by B7 family members are critical for both stimulating and inhibiting T-cell activation[1]. In conjunction with signaling through the T-cell receptor, CD28 ligation by B7 molecules stimulates antigen-specific proliferation of T cells and enhances production of cytokines, differentiation, and the effector function of T cells[2]. In contrast, B7 interaction with cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4, or CD152) delivers a negative signal that inhibits T-cell proliferation, IL-2 production, and cell cycle progression[3]. In addition to CTLA-4, several studies have indicated that PD-1, a member of the immunoglobulin (Ig) superfamily and a CD28 homolog, functions as an important negative regulator of the immune system[2]. The ligands of PD-1, PD-L1 (B7-H1), and PD-L2 (B7-DC) have recently been identified[4–6], which deliver an inhibitory signal by binding to the common receptor PD-1[4], although some studies suggest a stimulatory signal is delivered by the PD ligand (PD-L) through a putative receptor other than PD-1[2,5]. PD-L1 and PD-L2 are homologous to B7 molecules and belong to the Ig superfamily[2]. Despite their homology with B7 molecules, PD-L1 and PD-L2 do not bind to CD28 or CTLA-4[5,6], suggesting that they possess different roles from B7 molecules.
Because of their important roles in the immune response, the expression of the B7 family is strictly regulated. More-over, their expression is further modulated by alternative splicing of the transcripts[2]. Some of these alternatively spliced isoforms play an important part in the modulation of immune responses. Study on CTLA-4 isoforms, for instance, indicates an association of lower mRNA levels of the soluble alternative splice form with susceptibility to autoimmune disease[7]. Similarly, differential splice variants have also been identified for B7-1 and B7-2 genes and its receptor CD28[8–10]. It has been demonstrated that a soluble B7-2 is a costimulatory molecule for human T cells[9]. Recently, we have identified two alternatively spliced variants of human PD-L2[11], although their functional implications remain to be discovered.
The present study reports the identification of a novel splicing variant of human PD-L1 mRNA, which encodes an isoform lacking immunoglobulin variable domain (Igv)-like domain in the extracellular region. The expression and subcellular localization of this isoform was compared with the conventional product. Differential expression patterns of PD-L1 variants of different statuses suggest alternative splicing of PD-L1 may modulate the immune response in peripheral tissues.
Materials and methods
Cell preparation and activation To prepare peripheral blood mononuclear cells (PBMC), heparinized human peripheral blood was collected from two adult volunteers by venipuncture. Blood samples were diluted 1:2 with phosphate-buffered saline (PBS) and a mononuclear cell fraction was obtained by centrifugation over a Ficoll-Hypaque density gradient (Lymphoprep; NYCOMED, Elverum, Norway). PBMC were collected from the interface and washed twice with PBS and resuspended in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum, 10 mmol/L glutamine, 2-mercaptoethanol 50 μmol/L, benzylpenicillin 100 kU/L, and streptomycin 100 mg/L (Invitrogen, Carlsbad, CA, USA) at a final concentration of 2×109 cells/L. The cells were incubated in a humidified atmosphere with 5% CO2 for 48 h at 37 ºC in the presence of 1×10–7 mol/L phorbol 12,13-dibutyrate (PDB; Calbiochem, San Diego, CA, USA) and 0.5 µg/mL ionomycin (Sigma, St Louis, MO, USA) or 10 mg/L phytohemagglutinin (PHA; Sigma).
Reverse transcription polymerase chain reaction amplification (RT-PCR) of PD-L1 cDNA Heparinized human peripheral blood was collected from two donors by venipuncture. Total RNA was extracted from freshly isolated and activated PBMC using TRIzol reagent (Invitrogen). cDNA were synthesized from the isolated RNA using the ThermoScript RT-PCR system according to the recommended procedure (Invitrogen). RNA integrity was verified by both electrophoresis in 1% (w/v) agarose gels and PCR amplification of the cDNA of a house-keeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). PCR amplification of the resultant cDNA was performed in a total volume of 50 µL containing high fidelity DeepVent Taq polymerase (New England Biolabs, Beverly, MA, USA). For amplification of PD-L1, PCR was performed with an initial denaturation for 2 min at 94 ºC, followed by 35 cycles of 94 ºC for 30 s, 50 ºC for 30 s, and 72 ºC for 1 min, and a final extension for 10 min at 72 ºC. The primers (5'-CCG CGA ATT CAT GAG GAT ATT TGC TGT CTT TA-3' and 5'-ATC AGG TAC CTT ACG TCT CCT CCA AAT GTG TAT C-3') were designed to amplify the entire coding sequence of human PD-L1 cDNA[4]. For amplification of the GAPDH cDNA, PCR was performed as above, except that the annealing temperature was 60 ºC and the primers (5'-AAG GCT GAG AAC GGG AAG CTT GTC ATC AAT-3' and 5' -TTC CCG TCT AGC TCA GGG ATG ACC TTG CCC-3') were designed to amplify the target sequence. All primers were synthesized at BioAsia (Shanghai, China).
Cloning and sequencing of PD-L1 cDNA The PCR products were separated by electrophoresis in 1% (w/v) agarose gels containing ethidium bromide and recovered from the gel using a QIAquick Gel Extraction kit (QIAGEN, Hilden, Germany). The fragments of 873 bp and 532 bp were digested separately with EcoRI and KpnI, and cloned into a pEGFP-N1 vector (ClonTech, Palo Alto, CA, USA). Randomly selected transformed DH5α clones were screened with EcoRI and KpnI cutting for the presence of a correctly sized insert. Nucleotide sequences of independent clones from the samples of 2 donors were determined using dye-labeled deoxy-terminator, a 377 automated DNA sequencer (Applied Biosystem, Foster City, CA, USA).
Expression of PD-L1-EGFP fusion proteins in K562 cells PD-L1 type I or type II cDNA containing the sequence encoding signal peptide was separately subcloned into the mammalian expression vector pEGFP-N1 to construct the vectors for the fusion proteins of PD-L1 isoforms with enhanced green fluorescent protein (EGFP). These vectors were named pEGFP/PD-L1I (for the type I isoform) and pEGFP/PD-L1II (for the type II isoform). A Kozak sequence was included at the translational initiation region and the stop codon was deleted. EGFP was thus fused at the C-terminus of the PD-L1 isoform. For transient transfection, K562 cells were grown in RPMI-1640 supplemented with 10% (v/v) fetal bovine serum at 37 ºC in a CO2 incubator. Cells were transfected using a Cell Line Nucleofector kit V and Nucleofector Device (Amaxa, Cologne, Germany) according to the recommended protocol with 2 μg of the expression vector pEGFP/PD-L1I, pEGFP/PD-L1II, or empty vector pEGFP-N1, respectively, and left for 24 h in a CO2 incubator. These cells were subsequently stained with phycoerythrin (PE)-labeled mouse-antihuman PD-L1 monoclonal antibody (PE-PD-L1, e-Bioscience, San Diego, CA) and fixed with 4% (w/v) paraformaldehyde. The fluorescence of EGFP and PE was then analyzed by flow cytometry (FACSCalibur; Becton Dickinson, San Jose, CA, USA) and confocal microscopy (Spinning Disk Confocal Microscope; Perkin Elmer, Wellesley, MA, USA).
Western blot Cells (1×106) were dissolved in loading buffer and separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% w/v) according to Laemmli’s discontinuous system[12]. After electrophoresis, the gels were blotted onto nitrocellulose paper (Bio-Rad, Hercules, CA, USA) for Western blot analysis to visualize the specific protein bands. The Western blots were incubated with the goatantihuman PD-L1 antibody (R&D, Minneapolis, MN, USA). Horseradish peroxidase-conjugated rabbit-antigoat antibody (Jackson ImmuneResearch, West Grove, PA, USA) was used as the secondary reagent, and the color reaction was developed using 4-chloro-1-naphthol as a substrate. The molecular mass was evaluated with a software package (PhotoCapt Ver11.01; Vilber Lourmat, Marne La Vallée, France).
Bioinformatics Homology analyses of the cloned sequences were performed using a BLAST[13] search on the NCBI website and a PROSIS software package (Hitachi Software, Yokohama, Japan). The human genomic DNA sequence (submitted to the GenBank database under accession number AL162253) from clone RP11-574F11 on chromosome 9, which contains the PD-L1 gene, was used to analyze exon-intron junction sequences. The transmembrane regions were predicted using TMpred, a web-based pro- gram[14]. SignalP[15] was used to identify the potential signal peptide.
Results
Cloning and sequence analysis of a PD-L1 splice variant RT-PCR was performed to amplify the full coding sequence of PD-L1 mRNA with specific primers. PCR product resolution on agarose gel revealed that the product of activated PBMC had one main band of 873 bp and a minor band of 532 bp (Figure 1A, lane 3), whereas that of freshly isolated PBMC only had the shorter one (Figure 1A, lane 2). The fragments of 873 bp and 532 bp were inserted separately into the pEGFP-N1 vector. Nine independent transformed DH5a clones were identified to have the expected inserts. DNA sequencing of these clones showed that 5 clones contained the 873 bp DNA fragment, which was the complete coding sequence for conventional PD-L1 (designated PD-L1I, submitted to the GenBank database under accession number AY254342), and which was identical to the previously reported cDNA of human PD-L1 (submitted to the GenBank database under accession number AF233516[4]). The other 4 clones contained the 532 bp fragment and were identical to PD-L1 cDNA except for a deletion of a 341 bp fragment as revealed by BLAST analysis. Sequence homology searches performed with the BLASTn algorithm[13] revealed that this sequence might be an alternatively spliced variant of PD-L1 mRNA.
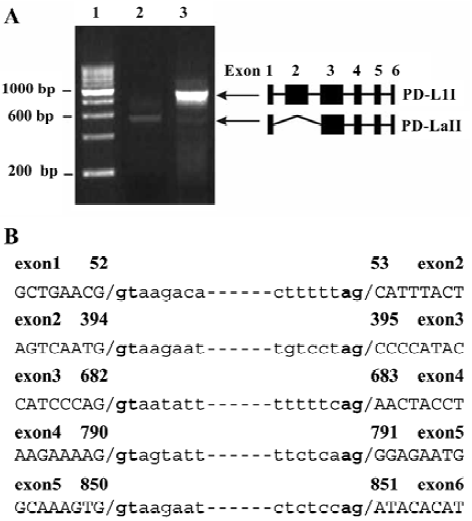
To examine how this variant was generated, the exon-intron junction sequences (Figure 1B) were analyzed based on the DNA sequence containing the PD-L1 gene (submitted to the GenBank database under accession number AL162253). The result showed that this variant was an alternatively spliced variant, which was produced by splicing out exon 2 to the canonical acceptor site at the 5’ end of exon3 while retaining all other exons (Figure 1A). This splice variant was designated PD-L1II (submitted to the GenBank database under accession number AY291313).
Analysis of amino acid sequence deduced from PD-L1 splice variant To analyze the putative translated protein of the PD-L1II, the deduced amino acid sequence was aligned with the conventional product PD-L1I using the PROSIS program. PD-L1I and PD-L1II variants contain open reading frames of 290 and 176 amino acid residues, respectively (Figure 2). Both of them contain the transmembrane domain as predicted with the TMpred program[14], and the signal peptide identified with the SignalP program[15], whereas the alternatively spliced isoform PD-L1II lacks an IgV-like domain but retains all other domains.

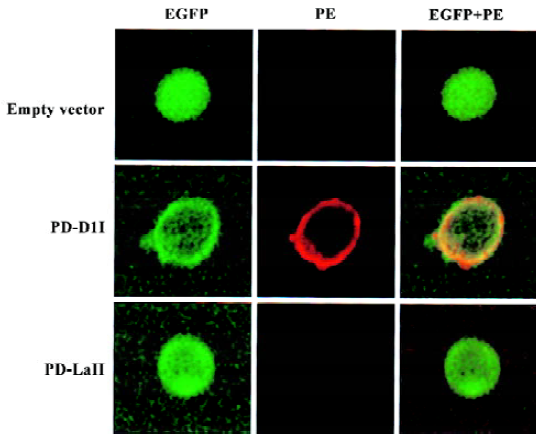
Expression and subcellular localization of the PD-L1II isoform fused with EGFP PD-L1 is a transmembrane protein, which would be expressed on the plasma surface, but direct visualization of its subcellular localization has not been reported. To determine whether the conventional product, PD-L1I, was located at the cell surface, and to investigate the distribution of PD-L1II, we examined the subcellular localization of their proteins in K562 cells transfected with pEGFP/PD-L1I or pEGFP/PD-L1II by flow cytometry and confocal microscopy. The expression of PD-L1I-EGFP and PD-L1II-EGFP fusion proteins was verified by Western blot (Figure 3). The estimated molecular masses of these two products were 74.3 kDa and 52.3 kDa, respectively, which were larger than the theoretical molecular masses of PD-L1I-EGFP (60 kDa) and PD-L1II-EGFP (47 kDa), probably because of posttranslational glycosylation. Thus, the glycosylation extent of PD-L1II was likely to be lower than that of PD-L1I. Moreover, flow cytometry analysis revealed that only the cells transfected with pEGFP/PD-L1I expressed PD-L1 on the cell surface, whereas no PD-L1 could be detected on the surface of the cells transfected with pEGFP/PD-L1II using antihuman PD-L1 antibody (Figure 4). To further investigate the subcellular distribution of the fusion proteins, confocal microscopy showed that PD-L1I-EGFP was largely located on the plasma membrane in the pEGFP/PD-L1I transfected cells, which could be stained by the specific monoclonal antibody for human PD-L1, indicating that it could be transported to the cell surface. Interestingly, PD-L1II-EGFP showed a pattern of intracellular membrane distribution as revealed by EGFP fluorescence, and those cells transfected with pEGFP/PD-L1II could not be stained by the specific antibody (Figure 5).


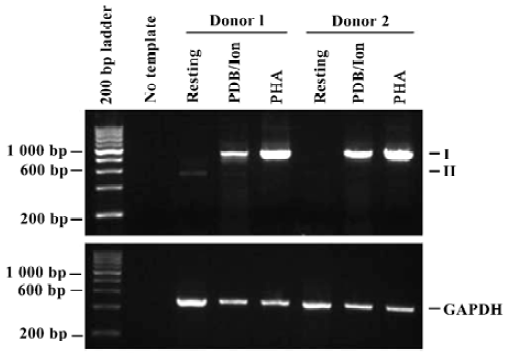
Differential expression of PD-L1 variants To compare the expression pattern of these PD-L1 mRNA variants in the resting and activated PBMC, total RNA of freshly isolated (resting) and activated PBMC of two unrelated donors were examined by RT-PCR (Figure 6). There was only the PD-L1II mRNA variant in the resting PBMC from donor 1, whereas both PD-L1I and PD-L1II variants existed in the PDB plus ionomycin-activated cells of the same donor, although the level of the PD-L1II variant was much lower. However, only the PD-L1I variant was detected in PHA-activated cells. In contrast, only the PD-L1I mRNA variant was amplified in the activated PBMC from donor 2 and there was a very low level of the PD-L1II mRNA variant in the resting cells from donor 2.
Discussion
Alternative splicing of costimulatory molecules is a common phenomenon and probably an important mechanism for regulating their function during immune responses. For instance, the splice variants of both CD28/CTLA-4 and their ligands B7-1/B7-2 have been identified, and these isoforms have been shown to play a role in regulating T-cell responses[7–10]. Recently, an alternative variant of PD-L2 that lacks an Igv-like domain has been identified in mice[6], and a human PD-L2 variant lacking an Igc-like domain has also been described[11]. However, no such splice variant of human PD-L1 has been reported to our knowledge. In the present study, we identified an alternatively spliced variant of PD-L1 mRNA encoding an isoform lacking the Igv-like domain. The novel variant was generated through splicing out exon 2, encoding an Igv-like domain but retaining all other exons without a frame-shift. The PD-L1II isoform was located at an intracellular compartment, which was likely to be the endoplasmic reticulum. In contrast, the conventional product of PD-L1 was located on the plasma membrane. Moreover, PD-L1II mRNA was expressed in resting PBMC but diminished or was absent in activated cells dependent on stimulating mitogens. Our results suggest that alternative splicing may be one of the mechanisms for modulating PD-L1 expression and function in peripheral blood cells.
As the Igv-like domain of PD-L1/PD-L2 is critical for binding to PD-1[2], any isoform of PD-1 ligands lacking the Igv-like domain does not bind to PD-1. For instance, Latchman et al found that an alternatively spliced variant of murine PD-L2, with the Igv-like exon being deleted, did not bind to PD-1-Ig, indicating that the Igv-like domain is necessary for ligand binding[6]. Moreover, molecular modeling and site-directed mutagenesis indicates that the binding sites of PD-L1/PD-L2 for PD-1 are largely located within the Igv-like domain[16]. Therefore, the novel PD-L1II isoform identified in this study would not bind to the receptor PD-1 because of the absence of the Igv-like domain, although direct measurement needs to be carried out. In addition, this isoform was located in the intracellular compartment and was unable to be transported onto the cell surface, suggesting that it would not function properly because only those expressed on the cell surface can interact with receptors. We have found similar results for the PD-L2 isoform lacking the Igc-like domain, which is also located mainly in intracellular compartments[11]. Why these isoforms could not be transported to the cell surface was unclear when the study was carried out, but this was probably a result of incorrect folding and/or defects in glycosylation of the PD-L1II isoform in the endoplasmic reticulum. The present study shows that the glycosylation extent of PD-L1II is lower than that of PD-L1I, suggesting that potential glycosylation sites on the Igv-like domain may be important for PD-L1 transportation to the cell surface.
PD-L1 and PD-L2 are two ligands for PD-1, a costimula-tory molecule that plays an inhibitory role in regulating T-cell activation in the periphery[4,6,17,18]. Their expression is thus modulated precisely during immune responses[2]. PD-L1 and PD-L2 share 40% amino acid identity and are more homologous to each other than to other ligands of the B7 family[2]. However, the expression pattern of these molecules is significantly broader than that of other B7 family ligands. The transcripts of both PD-L1 and PD-L2 were detected at high levels in the placenta and at low levels in the spleen, lymph nodes, and thymus, and were absent in the brain[4–6]. Expression of PD-L1 and PD-L2 in both lymphoid and nonlymphoid tissues suggests that the PD-1/PD-1 ligand pathway may modulate immune responses in secondary lymphoid organs as well as in peripheral sites. We[19] and other investigators[5] have found that the resting T cells do not express PD-L1, but mitogen activation of human T cells results in cell surface expression of PD-L1 in addition to PD-1. Similarly activated B cells express PD-L1 and PD-L2 in addition to PD-1[4,6,20]. This suggests that at the interface of T and B contact, antigen signals can be modulated bidirec-tionally through PD-1, thereby limiting T-cell-receptor and B-cell-receptor signaling after activation. However, the results presented here show that the PD-L1II splice variant can be detected by RT-PCR in the resting PBMC, suggesting that the mRNA transcripts of PD-L1 were actually produced in the resting cells, but may be alternatively spliced to generate a variant encoding the PD-L1II isoform. Because of its intracellular localization as revealed by an EGFP tag, the PD-L1II isoform could not be detected using surface staining. Thus, alternative splicing prevented PD-L1 from expression on the surface of the resting cells, thereby blocking its function. Although the expression of PD-L1II at the protein level in PBMC awaits confirmation, our results suggest that PD-L1 expression may be modulated at the posttranscriptional level through alternative splicing of its transcript.
To conclude, we identified an mRNA variant encoding a novel isoform of PD-L1, which was localized in the intracellular compartment, probably the endoplasmic reticulum. Our data also demonstrate the importance of the Igv-like domain of PD-L1 for targeting it to the cell surface.
References
- Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional inactivation: a costimulatory signaling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol 1989;7:445-80.
- Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol 2002;20:29-53.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459-65.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027-34.
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999;5:1365-9.
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-I and inhibits T cell activation. Nat Immunol 2001;2:261-8.
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003;423:506-11.
- Faas SJ, Giannoni MA, Mickle AP, Kiesecker CL, Reed DJ, Wu D, et al. Primary structure and functional characterization of a soluble, alternatively spliced form of B7-1. J Immunol 2000;164:6340-8.
- Jeannin P, Magistrelli G, Aubry JP, Caron G, Gauchat JF, Renno T, et al. Soluble CD86 is a costimulatory molecule for human T lymphocytes. Immunity 2000;13:303-12.
- Magistrelli G, Jeannin P, Elson G, Gauchat JF, Nguyen TN, Bonnefoy JY, et al. Identification of three alternatively spliced variants of human CD28 mRNA. Biochem Biophys Res Commun 1999;259:34-7.
- He XH, Liu Y, Xu LH, Zeng YY. Cloning and identification of two novel splice variants of human PD-L2. Acta Biochim Biophys Sin 2004;36:284-9.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680-5.
- National Center for Biotechnology Information [homepage on the Internet]. Bethesda: USA. National Library of Medicine; c2005 [updated 2005 Jan 19; cited 2004 Oct 13]. Available from: http://www.ncbi.nih.gov/BLAST
- Swiss EMBnet Node [homepage on the Internet]. Epalinges: Swiss Institute of Bioinformatics; [updated 2004 Dec 20; cited 2004 Oct 14]. TMpred Server; [about 2 screens]. Available from: http://www.ch.EMBnet.org/software/TMPRED_form.html
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004;340:783-95.
- Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med 2003;197:1083-91.
- Wiendl H, Mitsdoerffer M, Schneider D, Chen L, Lochmuller H, Melms A, et al. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: a novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J 2003;17:1892-4.
- He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D, et al. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol 2004;173:4919-28.
- He XH, Xu LH, Liu Y, Cai XC, Zeng YY. Cloning of the cDNA of human PD-L1 gene and the expression of its extracellular domain in Escherichia coli. Chin J Cell Mol Immun 2004;20:659-63.
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996;8:765-72.