
Down regulation of cyclooxygenase-2 is involved in delayed neuro-protec-tion by ischemic preconditioning in rats
Introduction
A short period of ischemia is known to increase tolerance of the brain to a subsequent prolonged ischemia by reducing brain infarction and apoptosis, this phenomenon is termed as ischemic preconditioning (IPC) or ischemic tolerance[1]. The neuroprotection of cerebral ischemic preconditioning is divided into acute and delayed effects[2]. Delayed neuroprotection occurs 24 h after IPC, then becomes manifest thereafter and lasts up to 7 d.
Recently, it has been reported that indomethacin, a nonselective inhibitor of cyclooxygenase (COX), applied 1 h prior to lipopolysaccharide (LPS) treatment abolished the LPS-induce delayed neuroprotection against focal cerebral ischemia and reperfusion injury[3]. The results suggested that PGs pathway or inflammatory pathway might be involved in triggering delayed neuroprotection by LPS[3]. It has also been reported that PGs increased to a high level in the brain tissues within hours following a short period of ischemia[4]. However it is still unknown whether a rise of PGs in the brain is involved in the delayed neuroprotection induced by IPC.
COX, also known as prostaglandin H2 synthase, is the rate-limiting enzyme in the metabolism of arachidonic acid into prostanoids (PGs and thromboxanes). COX-2, which is normally expressed in the neurons of the brain, is inducible in response to mitogen, endotoxin, and cytokines. Previous studies have demonstrated that focal cerebral ischemia induced strongly expressed COX-2 in neurons in penumbra and contributed significantly to enlargement of infarction[5]. Since IPC limits infarction induced by ischemia and reperfusion injury, it may prevent the up-regulation of COX-2 following focal cerebral ischemia and reperfusion insult. Recently it was shown that IPC stimulus reprogrammed the response of gene transcription to subsequent ischemia and reperfusion insult in mice. It was found that COX-2 showed lower expression of mRNA by IPC using a microarray analysis[6].
In the present study, the PGs pathway was examined to evaluate whether it is involved in triggering cerebral IPC, and effects of IPC on COX-2 expression were evaluated following focal cerebral ischemia and reperfusion.
Material and methods
Materials Indomethcin (Sigma-Aldrich, St Louit, MO, USA) was dissolved in Me2SO. The compound 2,3,7-triphenyltetrazolium chloride (TTC, Sigma-Aldrich, St Louis, USA) was dissolved in saline. TRI-REAGENT-LS extraction kit was purchased from Molecular Research Center, Inc (Cincinnati, USA). RNasin, dNTP, oligo (dT) 18 primer, and Taq DNA polymerase were obtained from Sangon Biotechnology Co (Shanghai, China). M-MuLV reverse transcriptase was from Fermentas Inc (Vilnius, Lithuania). The DNA Master SYBR Green I was from Roche Co (Mannheim, Germany). BCA protein assay kit and ECL chemiluminescence system were purchased from Pierce Co (Rockford, USA). Goat Polyclonal antibody specific for COX-2 was from Santa Cruz Biotechnology, Inc (Santa Cruz, USA, working concentration 1:500). Rat mono-antibody specific for beta-actin was from Sigma-Aldrich (St Louis, USA, working concentration 1:5000). Supersignal West Pico Trial Kit was from Pierce Co (Rockford, USA).
Animals and experiment design Male Sprague-Dawley rats weighing 220–250 g (the Shanghai Experimental Animal Center of Chinese Academy of Sciences) were used in this study. The rats were randomly divided into six groups (n=8 each). In the sham group (sham), the left common carotid artery was exposed, and the external carotid artery and its branches were isolated and coagulated without middle cerebral artery occlusion (MCAO). The rats of the ischemia and reperfusion group (I/R) were subjected to MCAO (90 min) and reperfusion (24 h). For the IPC group (IPC), IPC was applied 48 h before MCAO and reperfusion. For indomethacin group 1 (Indo1+IPC+I/R), the rats were treated with indomethacin (3 mg/kg ip) 1 h prior to IPC, and MCAO and reperfusion was applied 48 h later. For indomethacin group 2 (Indo2+IPC+I/R), the rats were treated with indomethacin (3 mg/kg ip) 1 h after IPC, and MCAO and reperfusion were applied 48 h later. The last group was used to exclude the effects of indomethacin itself on injuries of MCAO and reperfusion. The rats were treated with indomethacin (3 mg/kg ip) 48 h before MCAO and reperfusion (Indo+I/R).
COX-2 mRNA and protein expression Another series of experiments were performed. The rats were divided into four groups (n=6 each). In the sham operation group, the left common carotid artery was exposed, and the external carotid artery and its branches were isolated and coagulated without MCAO. The rats of the ischemia and reperfusion group were subjected to MCAO (90 min) and reperfusion (24 h). In the third group, IPC was applied 48 h before MCAO and reperfusion. In the last group, only IPC was applied.
Model of IPC IPC was induced as described previously with minor modification[7]. Briefly, the left common carotid artery was exposed, and the external carotid artery and its branches were isolated and coagulated. A branch of the left internal carotid artery (the left pterygopal artery) was isolated and coagulated. The right common carotid artery was exposed. Then a cannula was inserted into the external carotid artery stump and advanced into the internal carotid artery to infuse saline into the brain. The saline infusion rate was 12 mL/h, which was controlled by a Harvard apparatus compact infusion pump (USA). A surgery artery nip was used to clamp the right common carotid artery. To achieve marked neuroprotection, we chose 48 h after IPC as the time interval to apply ischemia and reperfusion insult.
Evaluation of infarct area Coronal sections of the brain (2 mm thick) were cut and immersed in a 1% solution of TTC. The stained slices were then fixed by immersion in phosphate-buffered 10% formaldehyde. The infarct area and hemispheric area of each section were photographed by a digital camera and quantitated using the public domain image processing and analysis program developed at the National Institute of Health, USA. The infarct area was expressed as percentage to the contralateral hemisphere.
Real-time reverse transcription PCR The expression of RNAs was determined by quantitative real-time RT-PCR using a Continuous Fluorescence Detector (MJ Research Incorporated, DNA Engine Opticn 2) with the DNA Master SYBR Green I. Animals were killed 24 h after reperfusion and their brains removed. A 4-mm-thick coronal brain slice was cut at the levels of the optic chiasm, and the infarcted cortex was dissected using the corpus callosum as a ventral landmark[8]. Total RNA was prepared from the samples with TRI-REAGENT-LS extraction kit according to manufacture’s instructions. Complementary DNA was created from RNA using TrueScript MMLV reverse transcriptase and oligo d(T)18 primers. A 0.2 µg amount of RNA was included in each reaction in a total volume of 20 µL. The reaction was performed at 42 ºC for 2 h. Thereafter the mix was diluted five-fold, and 2 µL was added to the PCR reaction mixture to yield a total volume of 20 µL. The COX-2 primers were forward, 5'-CCA TGT CAA AAC CGT GGT GAA TG-3'; reverse: 5'-ATG GGA GTT GGG CAG TCA TCA G-3', which result in a PCR product of 374 bp. The amplification reaction consisted of 35 cycles of denaturation (94 ºC, 30 s), annealing (64 ºC, 30 s), and elongation (72 ºC, 45 s). The beta-actin primers were forward, 5'AAG ATG ACC CAG ATC ATG TT3'; reverse: 5'TTA ATG TCA CGC ACG ATT T3', which resulted in a PCR product of 286 bp. The amplification reaction consisted of 35 cycles of denaturation (94 ºC, 30 s), annealing (56 ºC, 30 s), and elongation (72 ºC, 45 s). Cycle numbers (crossing points, when amplification starts its exponential phase) were used for statistical analysis. The lower the cycle number indicates the higher the amount of initial template. The PCR products were verified by agarose gel electrophoresis.
Western blot analysis A 4-mm-thick coronal brain slice was cut at the levels of the optic chiasm, and the infarcted cortex was dissected using the corpus callosum as a ventral landmark[8]. The isolated cortex was homogenized in ice-cold lysis buffer. The protein content was determined by BCA protein assay. Equal amounts of protein per lane (50 μg) were loaded onto an 8% polyacrylamide gel and separated by electrophoresis at 120 V. Proteins were then transferred to PVDF membrane at 25 mA for 90 min, and the membrane was blocked with 5% nonfat dry milk in 1X TBS, 0.1% Tween-20 at 25 ºC with gentle shaking, overnight. The PVDF membrane was then incubated with a goat polyclonal antibody specific for COX-2 (1:500) 1 h at 25 ºC followed by horseradish peroxidase-conjugated secondary antibody (rabbit anti-goat) for 1 h at 25 ºC. Antibody labeling was detected by Supersignal West Pico Trial Kit. After being stripped, the same PVDF membrane was incubated with mono-antibody specific for beta-actin (1:5000) as an internal control. Western blot results were quantified by density.
Data analysis All data were expressed as mean±SD. Differences between different groups were assessed by a one-way analysis of variance and Student-Newman-Keuls test. A value of P<0.05 was considered statistically significant.
Results
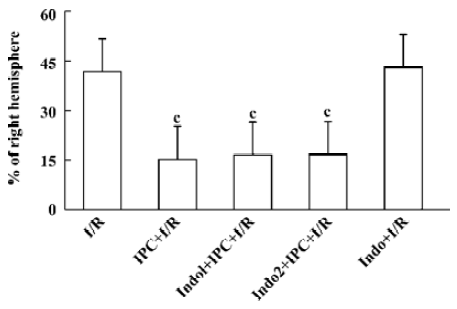
Protective effects of IPC At 24 h after operation, sham-operated rats did not show any cerebral tissue damage in TTC staining (not shown). IPC reduced the infarct area by 63% as compared with the MCAO alone group. Indomethacin (3 mg/kg ip), a nonselective inhibitor of COX, applied 1 h prior to or 1 h after IPC failed to affect these protective effects. Indomethacin itself had no effect on infarction by ischemia and reperfusion injury (Figure 1).
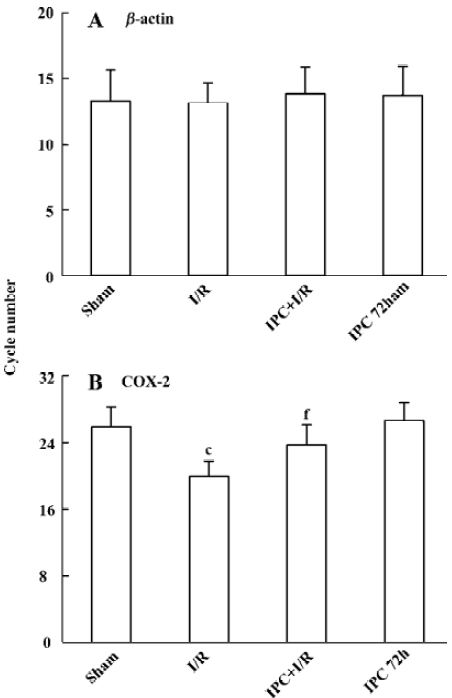
Effects of IPC on COX-2 mRNA expression In agreement with previous study, low levels of COX-2 PCR products were observed in the brain of sham-operated rats (to achieve the crossing points, the cycle number 25.8±2.4 was needed). After transient 90 min MCAO and 24 h reperfusion, COX-2 mRNA expression was significantly up regulated (the cycle number decreased to 19.9±1.9, P<0.01). IPC significantly prevented the up-regulation of COX-2 mRNA expression following ischemia and reperfusion injury (the cycle number decreased to 23.7±2.4, P<0.01), while IPC itself had no effect on the expression of COX-2 mRNA 72 h later (Figure 2).
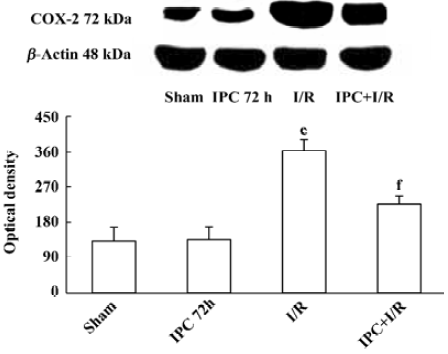
Effects of IPC on COX-2 protein expression COX-2 protein was expressed at a low level in the brain of sham-operated rats (132±35). IPC itself had no effects on expression of COX-2 protein 72 h later (135±32). Ischemia and reperfusion markedly increased COX-2 protein expression (362±28, P<0.01). IPC prevented the up regulation of COX-2 protein following ischemia and reperfusion injury (225±21, P<0.01) (Figure 3).
Discussion
IPC is a fundamental adaptive response of organisms against deleterious stress such as ischemia and other severe stimuli. In fact, this phenomenon exists in most organs such as brain, heart, skeletal muscle, lung, liver, stomach, intestine, and kidney. IPC shares some common mechanisms in different organs. The elements that constitute this molecular cascade of delayed IPC can be conceptually subdivided into three major components: (i) “triggers” or sensors, (ii) “effectors” or mediators, and (iii) “transducers”, signaling pathways that connect these two groups of molecules[1,9]. Endogenous active substances and bio-synthesized proteins have been suggested as triggers and end effectors of brain IPC, respectively. Adenosine, glutamate, oxygen free radical, and nitric oxide (NO) are these candidate substances that trigger brain IPC[1].
PGs are endogenous active substances that are suggested to be involved in IPC in the heart. It has been reported that pretreatment with indomethacin attenuated PGs release and abolished acute cardioprotection by IPC in different animal models in vivo or in vitro[10,11]. Exogenous PGs perfusion in isolated hearts mimicked cardioprotection induced by cardiac IPC[12]. The mechanisms of cardio-protection mediated by PGs are mainly related to the direct effects of PGs on the heart, including antagonism of adenylyl cyclase, activation of ATP-sensitive potassium channels, inhibition of calcium influx, and attenuation of neutrophil infiltration[13]. However, there was no study on the role of PGs in neuroprotection induced by IPC. A period of ischemia as short as 5 min can induce an early increase in PGs levels after 2 h of reperfusion in the brain[4]. However, cerebral IPC dose not seem to be triggered by increased PGs, since indomethacin failed to affect the protective effects of IPC. The negative role for PGs pathway may be attributed to the specific model of focal cerebral ischemia used for precondi-tioning. Saline infusion compromised oxygen and glucose supply, however, inflammatory response could be smaller, due to maintained idle perfusion at least partial being able to prevent from disturbed microcirculation and reduce leukocyte-endothelial cell adhesive interaction and so on. This in turn potentially diminished the inflammatory signal (including PGs pathway) as a trigger for preconditioning mechanisms. The result suggested the delayed neuroprotection by our model of IPC has different triggering mechanisms from that of delayed ischemic tolerance induced by LPS.
Three types of COX isoforms have been documented: COX-1, COX-2, and COX-3. COX-1 is constitutively expressed under physiological conditions. COX-3 is identical in sequence to COX-1 except for the in-frame retention of intron 1. Like its counterpart COX-1, COX-3 does not generally appear to be induced by acute inflammatory stimulation[14]. Several studies have demonstrated that COX-2 played an important role in the development of ischemic injury. In rodents as well as in humans, cerebral ischemia up-regulated COX-2 in neurons, blood vessels, and inflammatory cells in the injured brain[4,15,16]. Although COX-2 mRNA was not induced in the ischemic core, it was induced in the penumbral area in permanent or transient MCAO in rats. The up regulation of COX-2 began 6 h after ischemia, reached a maximum at 12–24 h and subsided at 48 h[4,8,17]. NS398, a selective COX-2 inhibitor, decreased infarction volume when administered 6 h after induction of ischemia[18]. In COX-2-deficient mice, there was a significant reduction in the MCAO-induced brain injury[19,20]. Our results that COX-2 mRNA and protein in the MCA cortex increased at 24 h after reperfusion in the rats of MCAO and reperfusion group were consistent with these previous studies. The mechanisms of the cytotoxicity of COX-2 induction may be related to the excessive release of arachidonic acid, and its products such as PGs, thromboxane and free oxygen radicals. These factors may act as the major inflammatory substances involved in the pathogenesis of ischemia and reperfusion[8]. COX-2 reaction products may also contribute to NMDA-induced neuronal injury and the pathogenesis of nitric oxide after ischemia[21,22].
Evidence has shown that COX-2 was an obligatory effector in delayed IPC in the heart[13]. Increased expression of COX-2 protein was observed 24 h after IPC with an increased enzymatic activity[23]. NS-398 and celecoxib, two selective inhibitors of COX-2, abolished the cardioprotection conferred by IPC [24]. It was also demonstrated that the expression of COX-2 subsided 48 h after a short period of ischemia in the brain[4,8,17]. Since the delayed neuroprotection produced by IPC lasts as long as 7 d and is much longer than that of delayed cardioprotection (72 h), the up-regulated COX-2 does not seem to be the effector of delayed neuro-protection by IPC in the brain. In the present study, we examined the expression of COX-2 after IPC or IPC followed by ischemia and reperfusion injury. Our results demonstrated that IPC had no direct effect on the cortex COX-2 mRNA and protein expression 72 h later. Thus, it is not likely that an up-regulation of COX-2 contributes to the delayed neuroprotec-tion by IPC in the brain. In the present study, we demonstrated that IPC significantly prevented the up-regulation of COX-2 mRNA and protein expression with a reduced infarction of the brain following ischemia and reperfusion injury. These results are similar to the previous finding that hyperbaric oxygen confers neuroprotection by depressing COX-2 expression and activity following ischemia and reperfusion injury[8]. So we speculate that while the up-regulation of COX-2 contributes to the delayed cardioprotection by IPC in the heart, it is probably that COX-2 takes an inverse role in delayed neuroprotection by IPC in the brain. Our results are consistent with the recent observation that COX-2 expression was down-regulated by ischemic preconditioning in the gerbil brain[25].
Recently, it has been shown that preconditioning stimulus reprogrammed the response of gene transcription to subsequent ischemia and reperfusion injury in mice. This feature mimics specific adaptive neuroprotective strategies seen in hibernation[6]. Depressed protein synthesis may be mediated in part via translational mechanisms that affect both initiation and elongation[6,26]. Since IPC confers robust neuroprotection, one presumption is that ischemic preconditioning may induce pro-survival factors that would negatively regulate COX-2. To date, three major mechanisms have been suggested to contribute to the neuroprotection by IPC, including anti-excitotoxic mechanisms, anti-apoptosis mechanisms, and anti-inflammatory mechanisms[1]. COX-2 has been verified to take an important role in pro-inflammatory action. We speculate that down-regulation of COX-2 may cooperate with other protective mechanisms and ultimately result in neuroprotection. However, more studies are needed to verify this hypothesis.
References
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003;26:248-54.
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab 1997;17:175-82.
- Bordet R, Deplanque D, Maboudou P, Puisieux F, Pu Q, Robin E, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab 2000;20:1190-6.
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al-Dalain S, Martinez G, et al. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem 2003;86:545-55.
- Koistinaho J, Chan PH. Spreading depression-induced cyclooxy-genase-2 expression in the cortex. Neurochem Res 2000;25:645-51.
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003;362:1028-37.
- Zhang FY, Cheng XC, Reng HM, Wu JS, Bao WM. A novel model of cerebral ischemic preconditioning in rats. Chin Clin Neurosci 2001;9:82-4.
- Yin W, Badr AE, Mychaskiw G, Zhang JH. Down regulation of COX-2 is involved in hyperbaric oxygen treatment in a rat transient focal cerebral ischemia model. Brain Res 2002;926:165-71.
- Bolli R. The late phase of preconditioning. Circ Res 2000;87:972-83.
- Song QJ, Xiao J, Deng HW, Li YJ. Role of calcitonin gene-related peptide in prostaglandins-mediated ischemic preconditioning in guinea pig hearts. Acta Pharmacol Sin 2001;22:3-9.
- Ma SG, Fu RF, Feng GQ, Wang ZJ, Ma XQ, Weng SA. Effect of G(alphaq/11) protein and ATP-sensitive potassium channels on prostaglandin E(1) preconditioning in rat hearts. Acta Pharmacol Sin 2004;25:587-92.
- Bouchard JF, Chouinard J, Lamontagne D. Participation of prostaglandin E2 in the endothelial protective effect of ischaemic preconditioning in isolated rat heart. Cardiovasc Res 2000;45:418-27.
- Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, et al. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res 2002;55:506-19.
- Shaftel SS, Olschowka JA, Hurley SD, Moore AH, O'Banion MK. COX-3: a plice variant of cyclooxygenase-1 in mouse neural tissue and cells. Brain Res Mol Brain Res 2003;119:213-5.
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in Neurons contributes to ischemic brain damage. J Neurosci 1997;17:2746-55.
- Ohtsuki T, Kitagawa K, Yamagata K, Mandai K, Mabuchi T, Matsushita K, et al. Induction of cyclooxygenase-2 mRNA in gerbil hippocampal neurons after transient forebrain ischemia. Brain Res 1996;736:353-6.
- Yokota C, Kaji T, Kuge Y, Inoue H, Tamaki N, Minematsu K. Temporal and topographic profiles of cyclooxygenase-2 expression during 24 h of focal brain ishemia in rats. Neurosci Lett 2004;357:219-22.
- Nagayama M, Niwa K, Nagayama T, Ross ME, Iadecola C. The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J Cereb Blood Flow Metab 1999;19:1213-9.
- Sasaki T, Kitagawa K, Yamagata K, Takemiya T, Tanaka S, Omura-Matsuoka E, et al. Amelioration of hippocampal neuronal damage after transient forebrain ischemia in cyclooxygenase-2-deficient mice. J Cereb Blood Flow Metab 2004;24:107-13.
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA 2001;98:1294-9.
- Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther 2000;293:417-25.
- Kittaka M, Giannotta SL, Zelman V, Correale JD, DeGiorgio CM, Weiss MH, et al. Attenuation of brain injury and reduction of neuron-specific enolase by nicardipine in systemic circulation following focal ischemia and reperfusion in a rat model. J Neurosurg 1997;87:731-7.
- Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol 2003;35:525-37.
- Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA 2000;97:10197-202.
- Colangelo V, Gordon WC, Mukherjee PK, Trivedi P, Ottino P. Downregulation of COX-2 and JNK expression after induction of ischemic tolerance in the gerbil brain. Brain Res 2004;1016:195-200.
- Frerichs KU, Smith CB, Brenner M, DeGracia DJ, Krause GS, Marrone L, et al. Suppression of protein synthesis in brain during hibernation involves inhibition of protein initiation and elongation. Proc Natl Acad Sci USA 1998;95:14511-6.