
Synthesis and anti-tumor activity of alkenyl camptothecin esters1
Introduction
Camptothecin, a pentacyclic alkaloid derixxved from a Chinese tree Camptotheca acuminata, showed good anti-tumor activity against the mouse leukemia L1210 system[1]. Camptothecin itself is water-insoluble. The sodium form of this compound was prepared and used in early clinical trials. Unfortunately, this carboxylate form of the molecule did not show the inherent anti-tumor activity, but severe toxicity[2–6], which caused the discontinuation of human clinical trials in the 1970s. Pharmacology and activity studies conducted since then have demonstrated that the lactone form and the 20-S-OH group of the compound are the most important structural requirements for anti-cancer activity. For example, the carboxylate sodium salt form showed only one-tenth the potency of the lactone form in an anti-tumor assay (P388 rodent leukemia) conducted by Wani et al[7]. In order to increase the stability of the lactone form of the molecule, we previously reported the preparation of alkyl camptothecin esters (Scheme 1)[8]. These alkyl camptothecin esters substantially increase the stability of the lactone form of the drug in mouse and human plasmas, and also the biological life of the active form of the drug in body. Zhao et al also reports that the 20-acylation of camptothecin brings lactone stabilization[9]. It is established that the 20-acylation of camptothecin is an effective method of increasing the lactone stability of the molecule.
Our structure-activity relationship (SAR) studies of alkyl camptothecin esters indicated that the anti-tumor activity of alkyl camptothecin esters A depends on the geometric shape of their side alkyl chains. For example, of all alkyl camptothecin esters tested, the compound with a C2 straight alkyl chain (R1=CH2CH3) shows good anti-tumor activity[10,11]. The compound is less active when R1 group in structure A becomes longer (C3, or C4), or bulkier (isopropyl group, or cyclic propyl group). The compound is inactive when the R1 group in structure A becomes larger (C5, C6, etc). Based on these SAR studies we propose that the introduction of an unsaturated functional group such as a double-bond or an epoxy group into the side ester chain in structure A would have an effect on anti-tumor activity. We now wish to report the preparation of alkenyl camptothecin esters 3, 4, 7, and 8 and the corresponding epoxy derivatives 5-6 together with the preliminary results of biological studies with these new com-pounds.
Materials and methods
General Dry nitrogen was routinely used as the reaction atmosphere in all reactions. All glassware was baked at 70±10 ºC for a minimum of 2 h before being used. Melting points were obtained with a MEL-TEMP melting point apparatus and were uncorrected. The 1H NMR spectrums of approximately 10% (w/v) solution in CDCl3 were obtained at 270.05 MHz with a JEOL GX-270 WB NMR spectrometer. Chemical shifts are reported in parts per million (δ scale), employing tetramethylsilane as an internal standard. In reporting the NMR data, we have used the following abbreviations: coupling constants in Hertz (J), singlet (s), doublet (d), triplet (t), multiplet (m), and etc. Mass Spectra were recorded using a VG ZAB-SEQ mass spectrometer (VG Analytical Co, England) with a resolution of 10 000. Routinely used solvents such as THF and methylene chloride were dried and freshly distilled. Silica gel (230–400 mesh, Aldrich) for column chromatography was used for all product separations. TLC sheets (Silica gel with fluorescent indicator on poly-ethylene) employed in TLC (Thin-layer Chromatography) operations were purchased from Aldrich Chemical Co (Aldrich, Milwaukee, WI). The numbering system used in reporting NMR data were shown in Scheme 2. Camptothecin was purchased from China and purified before being used. Nine-nitrocamptothecin was prepared in our laboratory by using an established procedure[14].
Preparation of camptothecin-20-O-crotonate 3 Crotonic anhydride (40 mL) and pyridine (30 mL) were mixed in a 100 mL round-bottomed flask equipped with a magnetic stirrer. To this solution, 8 g (0.0230 mol) of the starting camptothecin 1 was added. The mixture was stirred at 110±10 ºC with a sand bath for 15 h. After cooling to room temperature, the reaction mixture was poured onto 1000 mL petroleum ether while stirring. The crude products precipitated from petroleum ether were collected by filtration and then separated by column chromatography with THF-CH2Cl2 (1:15) as eluent. Pure product 3 (3 g) was obtained as white powder after precipitation from 300 mL of petroleum ether, mp 218–220 ºC (deformed at 155 ºC), yield 31%, purity 99% (HPLC); 1H NMR: δ; 1.00 (3H, t, J 7.51, C19-methyl protons), 1.94 (3H, dd, JH25-H24 6.96, JH25-H23 1.89 Hz, C25-methyl protons), 2.15–2.35 (2H, m, C18-methylene protons), 5.28 (2H, s, C5-methylene protons), 5.38–5.74 (2H, dd, J 17.22, 17.22, C17-methylene protons), 5.99 (1H, d, J 13.92, C23-H), 7.05–7.14 (1H, dq, JH24-H23 15.38, JH24-H25 6.96, C24-H), 7.24 (1H, s, C14-H), 7.67 (1H, t, J 6.96, C10-H), 7.83 (1H, t, J 6.96, C11-H), 7.94 (1H, d, J 7.70, C9-H), 8.21 (1H, d, J 8.43, C12-H), 8.39 (1H, s, C7-H); m/z (relative intensity) 416 (M+, 20), 330 [M-86 (CH3CH=CHCOOH), 100], 315 [M-101 (CH3CH=CHCOOH+CH3), 40)], 302 [M-114 (CH3CH=CHCOOH+CO), 73)], 287 [M-129(CH3CH= CHCOOH+CO2+H), 30], 274 (10), 259 (9), 246 (9), 234 (3), 218 (5), 205 (4), 191 (3). HMRS (C24H20N2O5): Found: 416.137, required, 416.137.
Preparation of 9-Nitrcamptothecin-20-O-crotonate 4 Using 3.0 g (0.0076 mol) of 9-nitrocamptothecin 2 as the starting material, the reaction was carried out in the same manner as when preparing 3. Pure product 4 (2.5 g) was obtained as yellow powder, mp 224–225 ºC (deformed twice at 180 ºC and 203 ºC respectively), yield 71%, purity 99% (HPLC); 1H NMR: δ; 1.01 (3H, t, J 7.48, C19-methyl protons), 1.94 (3H, dd, JH25-H24 7.00, JH25-H23 1.83, C25-methyl protons), 2.15–2.33 (2H, m, C18-methylene protons), 5.36 (2H, s, C5-methylene protons), 5.40–5.73 (2H, dd, J 7.20 and 17.20, C17-methylene protons), 6.01 (1H, d, J 13.93, C23-H), 7.08–7.24 (1H, dq, JH24-H23 15.35, JH24-H25 6.98, C24-H), 7.26 (1H, s, C14-H), 7.92 (1H, t, J 6.98, C11-H), 8.47–8.53 (2H, m, C10-H and C12-H), 9.26 (1H, s, C7-H); m/z (relative intensity) 461 (M+, 15), 375 [M-86 (C+ H3CH=CHCOOH), 100], 360 [M-101 (CH3CH=CHCOOH+CH3), 32], 347 [M-114 (CH3CH=CHCOOH+CO), 74], 332 [M-129 (CH3CH=CHCOOH+CO+CH3), 32], 319 (10), 302 (10), 286 (13), 274 (8), 216 (7). HMRS (C24H19N3O7): Found, 461.122, required, 461.122.
Preparation of camptothecin 20-O-(2´,3´-epoxy)-butyrate 5 To 50 mL benzene in a 100-mL round-bottomed flask equipped with a magnetic stirrer 0.16 g (~0.0004 mol) of ester 3 and 150 mg of m-chloroperoxybenzoic acid (57%–86%, Aldrich Chemical) were added. The mixture was stirred at room temperature for a week. The solvent was removed by a rotary evaporator. The residue was chromatographically separated with THF-methylene chloride (1:10) as eluent. Pure product 5 (0.12 g) was obtained as white powder after precipitation from 100 mL of petroleum ether, mp 210–213 ºC (decomposition, the crystals deformed at 175 ºC), yield 72%, purity 99% (HPLC); 1H NMR: δ; 0.99 (3H, t, J 7.51, C19-methyl protons), 1.90–1.94 (3H, dd, JH25-H24 6.96, JH25-H23 1.84, C25-methyl protons), 2.07–2.33 (2H, m, C18-methylene protons), 5.30 (2H, s, C5-methylene protons), 5.38–5.72 (2H, dd, J 17.59 and 17.95, C17-metnylene protons), 5.95-6.02 (1H, dq, JH23-H24 15.75, JH23-H25 1.83, C23-H), 7.04–7.12 (1H, dq, JH24-H25 6.59, JH24-H23 15.38, C24-H), 7.22 (1H, s, C14-H), 7.75-8.01 (4H, m, C9-C12 aromatic protons), 8.78 (1H, s, 7-H); m/z (relative intensity) 432 (M+, 28), 416 [M-16 (O), 12], 346 [M-86 (C4H6O2), 100], 331 [M-101 (C4H5O3), 53], 318 [M-114 (C4H6O2+CO), 75], 303 [M-129 (C4H5O3+CO), 54], 287 [M-145 (C4H5O3+CO2), 27], 275 (15), 259 (7), 246 (8), 231 (5), 218 (8), 205 (10), 191 (5). HRMS (C24H20N2O6): Found, 432.132, required, 432.132.
Preparation of 9-Nitrocamptothecin-20-O-(2´,3´-epoxy)-butyrate 6 Using 0.9 g (~0.0020 mol) of 4 as starting material, the reaction was carried out in the same manner as when preparing 5. Pure product 6 (0.10 g) was obtained as yellow powders after precipitation from 100 mL of petroleum ether, mp 216–218 ºC(decomposition, deformed at ~196 ºC), yield 10%, purity 99% (HPLC); 1H NMR: δ; 1.00 (3H, t, J 7.50, C19- methyl protons), 1.91–1.95 (3H, dd, JH25-H24 6.97, JH25-H23 1.85, C25-methyl protons), 2.05-2.31 (2H, m, C18-methylene protons), 5.31 (2H, s, C5-methylene protons), 5.39–5.74 (2H, dd, J 17.60 and 17.94, C17-methylene protons), 5.97–6.04 (1H, dq, JH23-H24 15.77, JH23- H25 1.81, C23-H), 7.02-7.14 (1H, dq, JH24-H23 15.40, JH24-H25 6.61, C24-H), 7.24 (1H, s, C14-H), 7.78–8.10 (3H, m, C10-H, C11-H, and C12-H), 9.25 (1H, s, C7- H); m/z (relative intensity) 477 (M+, 30), 461 [M-16 (O), 10], 391 [M-86 (C4H6O2), 100], 376 [M-101 (C4H6O3), 55], 363 [M-114 (C4H6O2+CO), 73], 348 [M-29 (C4H6O3+CO), 51], 332 [M-145 (C4H6O3+CO2), 26], 320 (13), 314 (6), 301 (7), 286 (5), 273 (7), 268 (9), 254 (4). HRMS (C24H18N3O8): Found, 477.122, required, 477.121.
Preparation of camptothecin-20-O-cinnamate 7 Camptothecin 4.0 g (0.0115 mol) was added to 23 g (in excess) of cinnamoyl chloride in a 100 mL round-bottomed flask equipped with a magnetic stirrer and a sand bath. The mixture was stirred at 110±10 ºC for 24 h. After cooling to 50 ºC, the reaction mixture was poured onto 400 mL petroleum ether portion by portion while stirring. The crude product was collected by filtration after precipitation from petroleum ether. The crude product was then added to 300 mL of acetone-petroleum ether mixture (100 mL acetone, 200 mL petroleum ether) and was refluxed for 1 h. The pure product was obtained as pale yellow crystals after precipitation from the acetone-petroleum ether solvent system, mp 268 ºC (deformed at 250 ºC), yield 80%, purity 99% (HPLC); 1H NMR(Me2SO-d6): δ; 0.97 (3H, t, J 7.325, C19-methylprotons), 2.09–2.26 (2H, m, C18-methylene protons), 5.27 (1H, s, C5-methylene protons), 5.53 (2H, s, C17-methylene protons), 6.86 (1H, d, J 16.381, C23-H), 7.04 (1H, s, C14-H), 7.442–7.813 (8H, m, C10-H, C11-14, C24-C30-Hs), 8.09 (2H, t (d+d), J 8.545 and 9.156, C9H and C12-H), 8.66 (1H, s, C7-H); 13C NMR (Me2SO-d6): δ; 7.68 (C19), 30.35 (C18), 50.30 (C5), 66.37 (C17), 75.87 (C20), 94.70 (C23), 116.64 (C14), 118.96, 127.75, 128.01, 128.53, 128.82, 128.92, 129.05, 129.84, 120.43, 131.09, 131.68, 133.69, 145.49, 146.04, 146.95, 147.81, 152.26, 156.63 (C2, C3, C6-C16a, C24-C30), 165.00,167.32 (C21,C22); m/z (relative intensity) 478 (M+, 26), 330 [M-148 (C6H5CH=CHCOOH), 100], 315 (42), 302 (63), 287 (12), 259 (8), 246 (7), 234 (5); HRMS (C29H22N2O5): Found: 478.138, required, 478.137.
Preparation of 9-nitrocamptothecin-20-O-cinnamate 8 Nine-nitrocamptothecin 1.4 g (50% pure, prepared in our laboratory) was added all in once to 6.0 g of cinnamoyl chloride in a 100 mL round-bottomed flask equipped with a magnetic stirrer and a sand bath. The mixture was stirred at 110±10 ºC for 24 h. After cooling to 50 ºC, the reaction mixture was poured onto 100 mL petroleum ether while stirr-ing. The crude products were precipitated from the solvent. After filtration the crude products were chromatographically separated with THF-methylene chloride (1:15) as an eluent. The pure product was obtained as yellow powders after precipitation from petroleum ether, mp 282 ºC, yield 21%, purity 99% (HPLC); 1H NMR: δ; 1.04 (3H, t, J 7.325, C19-methyl protons), 2.156–2.40 (2H, m, C18-methylene protons), 5.36 (2H, s, C5-methylene protons), 5.42–5.75 (2H, dd, J 17.09 and 17.70, C17-methylene protons), 6.57 (1H, d, J 5.87, C23-H), 7.25 (1H, s, C14-H), 7.40–7.55 (5H, m, C10-H, C11-H, C12-H, C27-H, and C29-H), 7.74 (1H, d, J 16.47, C24-H), 7.87 (1H, t, J 7.94, C28-H), 8.44–8.48 (2H, m, C26-H, C30-H), 9.245 (1H, s, C7-H); 13NMR: d; 7.64 (C19), 31.91 (C18), 50.20 (C5), 67.07 (C17), 75.80 (C20), 97.08 (C23), 116.12 (C14), 120.90, 121.56, 125.80, 127.37, 128.38, 128.54, 128.90, 130.94, 131.48, 133.85, 136.48, 144.50, 145.96, 147.46, 157.60 (C2, C3, C6-C16a, C25-C30), 167.323 (C21 or C22, one of them not found); m/z (relative intensity) 523 (M+, 12), 375 [M-148 (C6H5CH=CHCOOH), 100], 360 (30), 347 (71), 332 (30); HRMS (C29H21N3O7): Found, 523.124, required, 523.123.
Growth inhibition assay To assess the anti-proliferative activity of the various esters, identical cell cultures received equimolar concentrations of these esters and the cell number per ml was counted using the Trypan blue exclusion method. Cells were seeded at a concentration of 2.5×104 cells per well in a 48-microwell plate. Three such plates were seeded for end-point observation of cytotoxicity after 3, 5, and 7 d of treatment. Two concentrations of each test compound were used, one which was equal to the known cytotoxic level of the parent compound and one which was four times that amount. Stocks consisted of fine suspensions of esters in polyethylene glycol (PEG-400; Aldrich). Control cultures received only the carrier. The cell number was counted at time points of 72, 120, and 168 h, respectively. The targeted cells included 14 human malignant cell lines: CLO breast carcinoma, MDA 231 breast carcinoma, BRO melanoma, SCH melanoma, MiaPaCa2 pancreatic carcinoma, panc1 pancreatic carcinoma, HT 29 colon carcinoma, MCCN colon carcinoma, EFO27 ovarian carcinoma, 2774 ovarian carcinoma, DU145 prostate carcinoma, PC3 prostate carcinoma, DOY lung carcinoma, and SPA lung carcinoma.
HPLC procedure for purity analysis The purities of the ester products were determined by HPLC analysis. The procedure was previously reported[12].
In vivo toxicity and anti-tumor activity All the animal experiments were performed on nude Swiss mice of the NIH, high-fertility strain. They were bred and raised in our laboratory under strict pathogen-free conditions. For the in vivo toxicity and anti-tumor activity determination, a tumor xenograft growing in a nude mouse, approximately 1 cm3 in size, was surgically removed under sterile conditions, finely minced with iridectomy scissors, and suspended in cell culture medium at the ratio 1:10, v/v. One half of 1 mL of this suspension, containing about 50 mg of wet-weight tumor mince was subcutaneously inoculated on the upper half of the dorsal thorax of the mouse. Groups of four or five animals were used. The drug was finely suspended in cottonseed oil and then injected into the stomach cavity of the mouse through the anterior abdominal wall using a 26-gauge needle. The weekly schedule previously established for intragastric injection of 9NC was once a day, 5 on, and 2 off. This schedule was employed throughout all the animal experiments. Treatment was initiated when the tumor had reached a volume of approximately 200 mm3, well-vascularized, measurable, and growing exponentially. Tumors growing in animals were checked and measured with a caliper once a week.
Statistical analysis Data were expressed as mean±SD and compared with t-test.
Results and discussion
Chemistry A number of published reports have shown the methodologies for 20-OH acylation of camptothecins. Wall et al successfully prepared Camptothecin acetate by using the straight acylation reaction of camptothecin with acetic anhydride in pyridine[1]. This compound was also prepared by two other different methods, one using acetic acid (rather than the reactive anhydride) as acylating agent with nitronium tetrafluoroborate as the catalyst, and one using trichloroethane as acylating agent with concentrate sulfuric acid as the reaction solvent[12,13]. We used crotonic anhydride as acylating reagent to prepare alkenyl camptothecin esters 3 and 4. Camptothein 1 and 9-nitrocamptothecin 2 were allowed to react with a largely excessive amount of crotonic anhydride in pyridine to give the corresponding products 3 (31% yield) and 4 (71% yield), respectively. The reaction mixture was heated with a sand bath at 110±10 ºC for 15 h. The products 3 and 4 were obtained respectively as white crystals (3) and yellow crystals (4) after separation by column and precipitation from petroleum ether. The subsequent epoxidation of 3 and 4 with m-chloroperoxybenzioc acid in benzene solvent gave the corresponding products 5 and 6 in yields of 70% and 10%, respectively (Scheme 2). Esters 7 and 8 were obtained in yields of 80% and 21%, respectively by heating the starting 1 or 2 in cinnamoyl chloride. The proton NMR spectra showed characteristic peaks at 1.00±0.05 ppm for C19-methyl protons of compounds. The C18-methylene protons of these compounds absorbed in the 2.05 to 2.35 ppm region with a multiplet peaks. The C25-methyl protons showed characteristic double doublets at 1.90±1.94 ppm for compounds 3–6. The alkenyl C23 proton of ester 3 and 4 showed characteristic peaks at 6.00±0.01 ppm; the other alkenyl C24 proton absorbed in the 7.04 to 7.24 ppm region. Esters 7 and 8 absorbed at 6.72±0.15 ppm for C23 proton. The C23 proton of esters 5 and 6 absorbed in the 5.95 to 6.04 ppm region with dq peaks because of the remote coupling between C23-proton and C25-methyl protons; the C24 proton of these two epoxy derivatives absorbed in the 7.02 to 7.14 ppm region. The mass spectra of these esters showed characteristic fragmentation pathway as depicted in Scheme 3.
Biological studies Cytotoxicity of these esters was observed in vitro by microwell plate screening as a first means of determining whether or not a compound warrants further evaluation. Standard microscopic observation of toxicity includes changes in morphology and the percent of cells remaining after specified amount of time in treated versus untreated cells. These esters were tested in vitro against 14 human malignant cell lines for anti-tumor activity. The experimental results were shown in Table 1. Table 2 showed the comparison of IC50 values of esters 3 and 4 to the commercially available comptothecin analogues, irinotecan (CPT-11) and topotecan. Figure 1 showed the anti-tumor activity of ester 3 against human breast carcinoma growing in nude mice as xenografts. Figure 2 showed the corresponding toxicity of ester 3 in mice. All esters tested in this study showed anti-tumor activity. However, larger doses were needed for these compounds in order to show the same strength of anti-tumor activity of their parent compounds, implying these esters are prodrugs. It is apparent from Table 1 that esters 3 and 4 are active against the tested cell lines and have almost the same strengths. It is also clear that from these tests that the activity of ester 5 is slightly higher than esters 3 and 4. Finally, the data in Table 1 showed that esters 6, 7, and 8 were only slightly active against those tested cell lines when comparing them to the control. We chose esters 3 and 4 for the further IC50 comparison studies because of the availability of the substances due to their easier syntheses. Irinotecan and topotecan are two camptothecin derivatives approved by FDA for treating cancer patients. The IC50 values of esters 3 and 4 are compared to the IC50 values of two marketed comptothecin analogues as shown in Table 2. In terms of IC50 values, ester 3 is 3 to 12 folds more potent than irinotecan against all testing cell lines and 1.3 to 3 folds more potent than topotecan against almost every testing cell line except for cell line McCain; ester 4 is 4 to 27 folds more potent than irinotecan and 1 to 4 folds more potent than topotecan against all testing cell lines. We also found that the anti-tumor activity of these new esters is same as or slightly higher than the corresponding alkyl derivatives when comparing the data in Table 1 to the data we previously reported for the corresponding alkyl ester compounds[10,11]. Thus, the introduction of an unsaturated double bond into the side ester chain of the molecule does not significantly alter the anti-tumor activity. We also chose ester 3 for the in vivo evaluation because of the availability of the larger scale synthesis of the molecule. Figure 1 shows the anti-tumor activity of ester 3 at a dose of 8 mg/kg against human breast carcinoma growing as xenografts in nude mice, and Figure 2 shows the corresponding toxicity of 3 at same dose level in mice. It is apparent form these in vivo studies that compound 3 completely inhibits the growth of human breast tumor in mice with no signs of toxicity as the bodyweight of mice increased slightly during the treatment.
In conclusion, alkenyl campothecin esters 3 and 4 can be obtained by the straight esterification of the corresponding starting mother compounds with crotonic anhydride. These two compounds are more potent than the commercially available camptoxthecin analogues, irinotecan and topotecan, in terms of IC50 values. Furthermore, compound 3 shows strong in vivo anti-tumor activity against human breast carcinoma in mice with no signs of toxicity. Thus, these alkenyl camptothecin esters have great potential to be developed for human treatment.
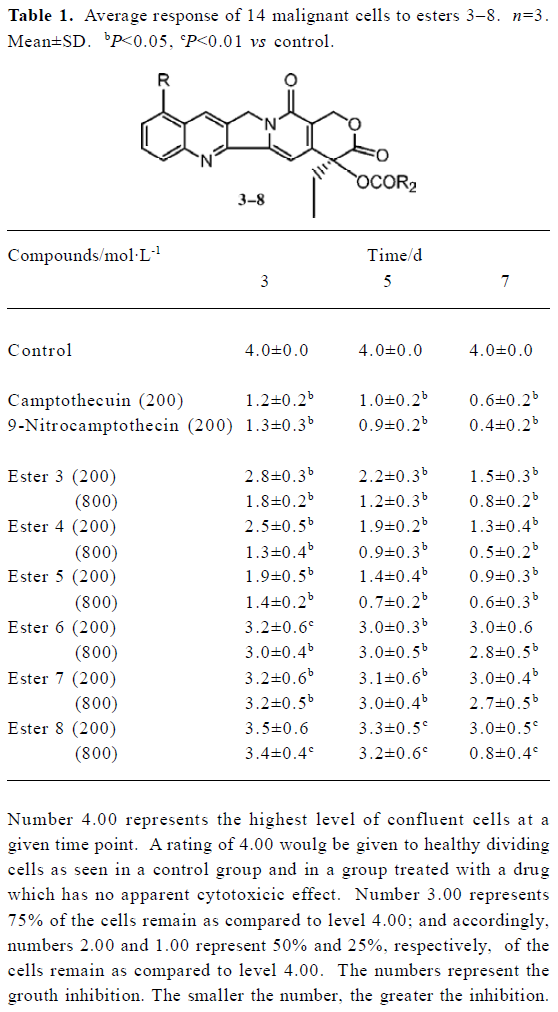
Full table
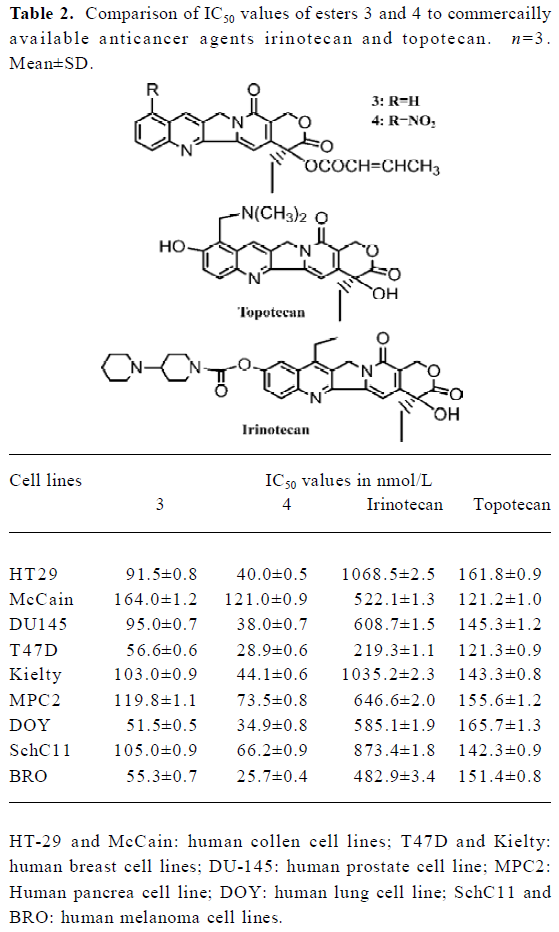
Full table
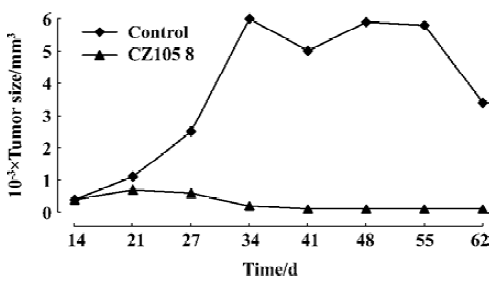
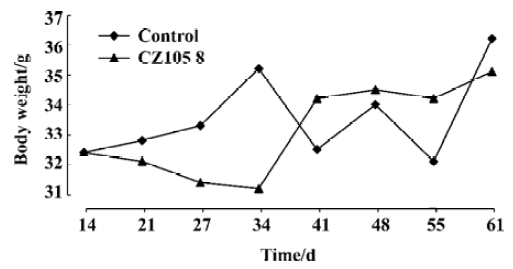
Acknowledgement
The friends of the Stehlin Foundation are gratefully acknowledged.
Footnote
Financial support from the Stehlin Founda-tion for Cancer Research.
References
- Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata. J Am Chem Soc 1966;88:3888-90.
- Gottlieb JA, Guarino AM, Call JB, Oliverio VT, Block JB. Preliminary pharmacologic and clinical evaluation of camptothecin sodium (NSC-100880). Cancer Chemother Rep Part 1 1970;54:461-70.
- Gottlieb JA, Luce JK. Treatment of malignant melanoma with camptothecin (NSC-100880). Cancer Chemother Rep 1972;56:103-5.
- Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Selawry OS. Phase I clinical trial of weekly and daily treatment with camptothecin (NSC-100880): correlation with preclinical studies. Cancer Chemother Rep 1972;56:515-21.
- Moertel CG, Schutt AJ, Reitemeier RJ, Hahn RG. Phase II studies of camptothecin (NSC-100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep 1972;56:95-101.
- Schaeppi U, Fleishman RW, Cooney DA. Toxicity of camptothecin (NSC-100880). Cancer Chemother Rep 1974;5:25-36.
- Wani MC, Ronman PE, Lindley LT, Wall ME. Plant antitumor agents. 18. Synthesis and biological activity of camptothecin analogs. J Med Chem 1980;23:554-60.
- Cao Z, Harris N, Kozielski A, Vardeman D, Stehlin J, Giovanella B. Alkyl esters of camptothecin and 9-nitrocamptothecin: synthesis, in vitro pharmacokinetics, toxicity, and antitumor activity. J Med Chem 1998;41:31-7.
- Zhao H, Lee C, Sai P, Choe Y, Boro M, Pendri A, et al. 20-O-Acylcamptothecin derivatives: evidence for lactone stablization. J Org Chem 2000;65:4601-6.
- Cao Z, Mendoza J, Early J, Harris N, Kozieski A, Liehr J, et al. Structure-activity relationship of alkyl camptothecin esters. The Camptothecins: unfolding their anticancer potential. Ann NY Acad Sci 2000;922:122-35.
- Cao Z, Mendoza J, Early J, Harris N, Kozieski A, Liehr J, et al. Structure-activity relationship of alkyl 9-nitrocamptothecin esters. Acta Pharmacol Sin 2003;24:109-19.
- Cao Z, Armstrong K, Shaw M, Petry E, Harris N. Nitration of camptothecin with various inorganic nitrate salts in concentrated surfuric acid: a new preparation of anticancer drug 9-nitrocampto-thecin. Synthesis 1998.1724-30.
- Cao Z. Preparation of 14-nitrocamptothecin derivatives by reactions of camptothecin with nitronium tetrofluoroborate in acidic solvents. J Chem Soc Perkin Trans 1 1996.2629-32.
- Cao Z. An alternative preparation of camptothecin 20(S)-acetate. Synth Commun 1997;27:2013-9.