
RNA interference by expression of short hairpin RNAs suppresses bcl-xL gene expression in nasopharyngeal carcinoma cells1
Introduction
RNA interference (RNAi) is a ubiquitous mechanism of eukaryotic gene regulation that can be exploited for specific gene silencing. In 1998, Fire and his colleagues firstly named these phenomena as RNAi[1]. Recently, RNAi has emerged as a powerful reverse genetic tool to silence gene expression in multiple organisms including plants, Caenorhabditis elegans, and Drosophila[2].
The specificity of RNAi is determined by 21- to 23-nt RNA duplexes, referred to as micro-RNAs (miRNA) or small interfering RNAs (siRNA). miRNAs are generated from endogenous precursors, which form hairpin structures with stretches of double-stranded RNA (dsRNA). They are cleaved by the ribonuclease Dicer to produce mature miRNA. After unwinding, one of the strands becomes incorporated into the RNA-induced silencing complex (RISC) and guides the destruction or repression of complementary mRNA. siRNA arise from viral or other exogenous dsRNA, but they use the same mechanism to effect mRNA degradation[3].
Nasopharyngeal carcinoma (NPC) is a gene-related malignancy that threatens many people’s lives in Southern China. Since its discovery, many people have lost their lives to this malignancy because of the lack of ideal therapeutics. Presently, overexpression of bcl-xL is widely considered as a poor prognosis in this cancer. In recent years, we have done much work to investigate its ideal therapeutics, including antisense gene therapy strategy. We found a favorable selectivity and high specificity of antisense RNA to treat NPC in vivo; however, the quick degradation of antisense RNA in cells and the unspecific toxicity limit its applications. RNA interference, a tool for inhibiting the overexpression of specific oncogenes, has been included in therapeutic studies of cancers in recent years[4].
One obstacle to achieve RNAi in mammals is that dsRNA longer than 30 nt will provoke an antiviral response, leading to the non-specific degradation of RNA transcripts and a global shutdown of host cell protein translation, known as a strong cytotoxic response. As a result, the long dsRNA does not produce RNAi activity as expected. This obstacle has recently been solved by Elbashir et al who found that gene specific suppression in mammalian cells can be achieved by vitro-synthesized siRNA that are 21 nt in length, long enough to induce gene-specific suppression, but short enough to evade the host interferon response[2,5]. However, this gene-specific suppression is transient, which severely restricts its applications. To overcome this restriction, various kinds of vectors have been designed to express siRNA or hairpin RNA transcripts to suppress expression of endogenous genes in mammalian cells[6–8].
In this article, we constructed a plasmid to achieve RNAi in mammalian cells. With this plasmid, shRNA are predicted to be synthesized from a DNA template under the control of mU6 promoter (a kind of eukaryotic RNA polymerase III promoter) in transfected cells. This construction has two BbsI cloning sites under mU6 promoter to ensure the coding sequences for shRNA insert in the right direction and direct the synthesis of shRNA in cells. Also, this plasmid containing a neomycin resistance gene (neor) can be used for positive screening. These properties make it possible to use DNA templates to synthesize shRNA in vivo and produce RNAi activity to trigger silencing of homologous gene expression. Using this plasmid, we found that bcl-xL in NPC cells can be efficiently inhibited and then induce apoptosis in NPC cells.
Materials and methods
Construction of plasmid containing DNA template for synthesis of shRNA under the control of mU6 promoter mU6 promoter was obtained by cutting mU6pro (gift of Li-ping LI, Guangdong Medical College, Zhanjiang, China) with BbsI and cloned into pcDNA3 instead of its CMV promoter to generate the plasmid pmU6. A general strategy for constructing an RNAi plasmid involved cloning an inverted repeat into pmU6 at the BbsI site. The selection of the coding sequences for siRNA were empirically determined but were analyzed by BLAST search to ensure that they did not have significant sequence homology with other genes. To generate the pmU6 bcl-xL RNAi plasmid, a 21-nt oligo corresponding to nucleotides (605–625) of the bcl-xL coding region was designed together with the 9-nt spacer, five Ts and two BbsI sites. They were then inserted into the pmU6 vector digested with BbsI to generate pmU6 bcl-xL RNAi plasmid. This oligonucletide sequence encoding shRNA for the targeted gene bcl-xL coding region was taken from GenBank accession Z23115 L20121, GI-510900 (nucleotides 605–625)[9,10]. Primitive oligo is 5´-GGAGATGCAGGTATTG-GTGAG-3´ (forward) and 5´-CTCACCAATACCTGCATC-TCC-3´ (reverse). Modified oligos are primitive oligos plus the 9-nt spacer, five Ts and two BbsI sites, Modified oligo is 5´ TTTGGGA GATGCAGGTATTGGTGAGTTCAAGAGA-CTCACCAATA-CCTGCATCTCCTTTTT 3´ (forward) and 5´ GCTAAAAA-AGGAGATGCAGGTATTGGTGA-GTCTCTTGAACTCAC-CAATACCTGCATCTCC 3´ (reverse).
Cell culture and transfections NPC cell line CNE-2Z cells were cultured in RPMI-1640 (GIBCO, NY, USA) complete culture medium which supplemented with 10% of heat-inactivated FBS (GIBCO, NY, USA), benzylpenicillin (100 kU/L) and streptomycin (100 mg/L) at 37 ºC in an incubator in a humidified atmosphere, with 5% CO2 in air. The cells were routinely passaged every 1 or 2 d[11]. For transfection, 0.5×105 cells mixed with 500 µL RPMI-1640 (Invitrogen, Carlsbad, CA,USA, supplemented with 10% of heat-inactivated FBS, without any antibiotic) were seeded into a well of a 24-well plate and incubated at 37 ºC for 24 h. pmU6-RNAi plasmid and pTR-UF5 plasmid (gift of Dr Muzyczka, Florida University, USA) harboring green fluorescence protein (GFP) reporter gene were co-transfected using a Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA)-mediated method according to the manufacturer’s protocol. Nearly confluent cells in 24-well plates were transfected with 2.0 µg of pmU6-RNAi plasmid and 0.2 µg of pTR-UF5 per well, the final concentration of plasmid was 0.4 µg/mL; the ratio of plasmid to lipofectamine was 1:1. After a 6 h-transfection, the medium was replaced by complete culture medium. Cells were continuously cultured untill harvest for analysis.
Transfection efficiency assay The procedure of transfection was the same as described above. Cells were harvested after 24 h by centrifugation for 5 min at 200×g, to remove cell debris and washed three times with phosphate buffered saline (PBS) by centrifugation for 5 min at 1000×g to remove culture medium. Cells were then suspended again in PBS again for analysis by flow cytometry (Coulter, Becton Dickinson, USA). For each samples, at least 1×104 cells were analyzed by flow cytometry. Results were presented as the number of green fluorescence cells versus the total cells detected by flow cytometry and repeated for at least 3 times[12] and analyzed by Student’s t-test.
Hoechst 33258 fluorescence staining CNE-2Z cells (1×105) were seeded in a T-10 cm2 cell culture plate and cultured in RPMI-1640 supplemented with 10% heat-inactivated FBS without antibiotic. After an incubation at 37 °C for 24 h, cells were transfected with 4.0 µg of pmU6-RNAi plasmid alone per plate. After a transfection for 6 h, the medium was changed to complete culture medium. Cells were collected 12 to 24 h after the transfection and centrifuged for 5 min at 200×g to remove cell debris, and cells were washed three time with 0.9% saline after a centrifugation for 5 min at 1000×g to remove culture medium. And then 10 µL of cell suspensions were treated with 10 µL of 10 mg/L Hoechst 33258 for 30 min in the dark. Ten µL of the stained cell suspensions were taken out and dripped on the slides and covered with a cover slip. The morphological changes of nuclei in CNE-2Z cells were observed by fluorescence microscope (×400) to discriminate native cells, apoptotic cells and necrotic cells. Cells were photographed and processed by Adobe PHOTOSHOP software.
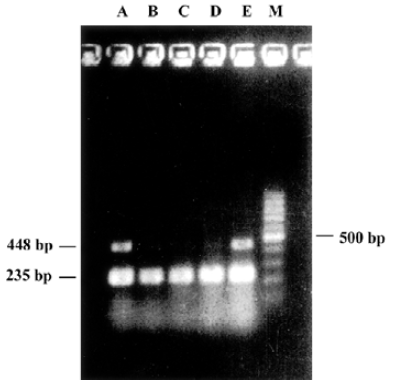
Quantitative RT-PCR Cells were collected 12 to 48 h after transfection and washed with PBS (pH 7.3), homogenized once and centrifuged at 1000×g for 5 min to remove culture medium. One mL TRIzol (Gibco, NY, USA) was added to the cell pellets, total RNA was extracted from with or without pmU6-RNAi plasmid transfected CNE-2Z cells according to a single step method (One-Step RT-PCR Kit, Applied QIAGEN Inc, USA). After centrifugation, the supernatant was extracted once with 0.2 mL chloroform and 0.5 mL isopropanol respectively, then total RNA was precipitated with 75% ice-cold ethanol (diluted with DEPC water). The precipitate was centrifuged and dissolved with 100 µL RNase-free water, after purification by a LiCl precipitating method, UV spectrophotometer analysis at 260 nm and electrophoresis detection showed good quality of purified RNA[13]. The upperstream primer of bcl-xL (amplified products were 448 bp) was 5´-CCC AGA AAG GAT ACA GCT GG-3´, the downstream primer was 5´-GCG ATC CGA CTC ACC AAT AC-3´. The expression of house keeping gene, β-actin mRNA, was used as an internal standard[14]. The upperstream primer of β-actin (amplified products were 235 bp) is 5´-GGG AGC CAA AAG GGT CAT CAT CT-3´, the downstream primer was 5´-GAG GGG CCA TCC ACA GTC TTC T-3´ (primers were commercially available). PCR were run 30 cycles for bcl-xL and β-actin in Eppendorf Mastercycler. Denaturing, annealing, and extension reactions were performed at 94 ºC for 45 s, 55 ºC for 1 min, and 72 ºC for 1 min. The PCR pro-ducts were electrophoresed on 1% agarose gel, stained with ethidium bromide, and detected by UV irradiation. The levels of bcl-xL mRNA were expressed as the ratio of bcl-xL to β-actin.
Western blotting The cells were washed three times with PBS and collected by scraping and were lysed in150 µL of ice-cold Tris buffer (50 mmol/L, pH 7.5) containing edetic acid 5 mmol/L, NaCl 150 mmol/L, 0.1% NP-40, 0.1% SDS, 2.0 g/L aprotinin, 0.02% NaN3, PMSF 0.2 mmol/L, and antiprotease mixture, sonicated, and centrifuged at 12 000×g for 15 min and the concentration of protein in each lysate was determined with Coomassie brilliant blue G-250. Loading buffer was added to each lysate, which was subsequently boiled for 3 min and then electrophoresed on a SDS-PAGE[15]. Before electrophoresis, the proteins were mixed with 2×loading buffer (containing Tris-HCl 100 mmol pH 6.8, 20% Glycerin, 4% SDS, tetrabromophenol-sulfonphthalein 0.05 g/L and 10% 2-β-mercaptoethanol) by the same volume. Proteins were transferred onto nitrocellulose and incubated with antibody and then with peroxidase-conjugated secondary antibody in the second reaction (The anti-bcl-xL antibody was used at a 1:500 dilution, the anti-β-actin antibody was used at a 1:400 dilution, the anti-HA mAb was used at a 1:2500 dilution, all antibodies was obtained from Peking Zhong-shan Biote-chnology, Beijing, China). Detection was performed with enhanced chemiluminescence agent. The electrochemi-luminescence (ECL) Test Kit-based detection was performed with Chemiluminescence Reagent (Sigma Life Science) according to the manufacturer’s instructions. The results of Western blot analysis represented the average of three individual experiments.
Results analysis and statistics The electrophoregram of RT-PCR and Western blotting were scanned by Scanner and the optical density scores of bands were calculated to analyze the relative content of bcl-xL to β-actin. Differences between individual groups were analyzed by One-Way ANOVA with SPSS10.0 software, and the results was expressed as mean±SD.
Results
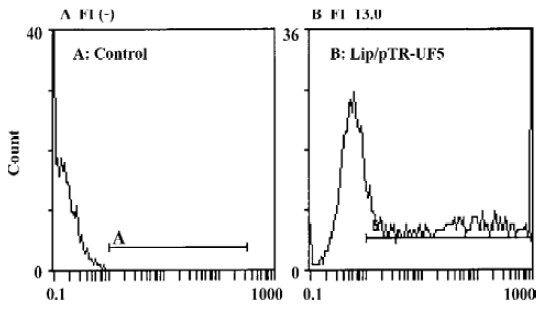
Analysis of transfection efficiency The expression of GFP in cells was detected by flow cytometry 24 h after co-transfection. Transfection efficiency was the rate of GFP expressing cells to the totally assayed cells; fluorescence index (FI) reflects the expression efficiency of the GFP gene[16]. The transfection efficiency of the untreated control group and pTR-UF5 group was (0.4±0.4)% and (50.7±4.4)% respectively, implying lipofectamine-2000 can efficiently introduce plasmid into cancer cells. The FI of the control group and pTR-UF5 group was negative (–) and 13.0±0.3 (P<0.01 vs control), respectively, showing GFP had a higher expression in NPC cells after introducing pTR-UF5 into cancer cells by lipofectamine-2000 (Figure 1).
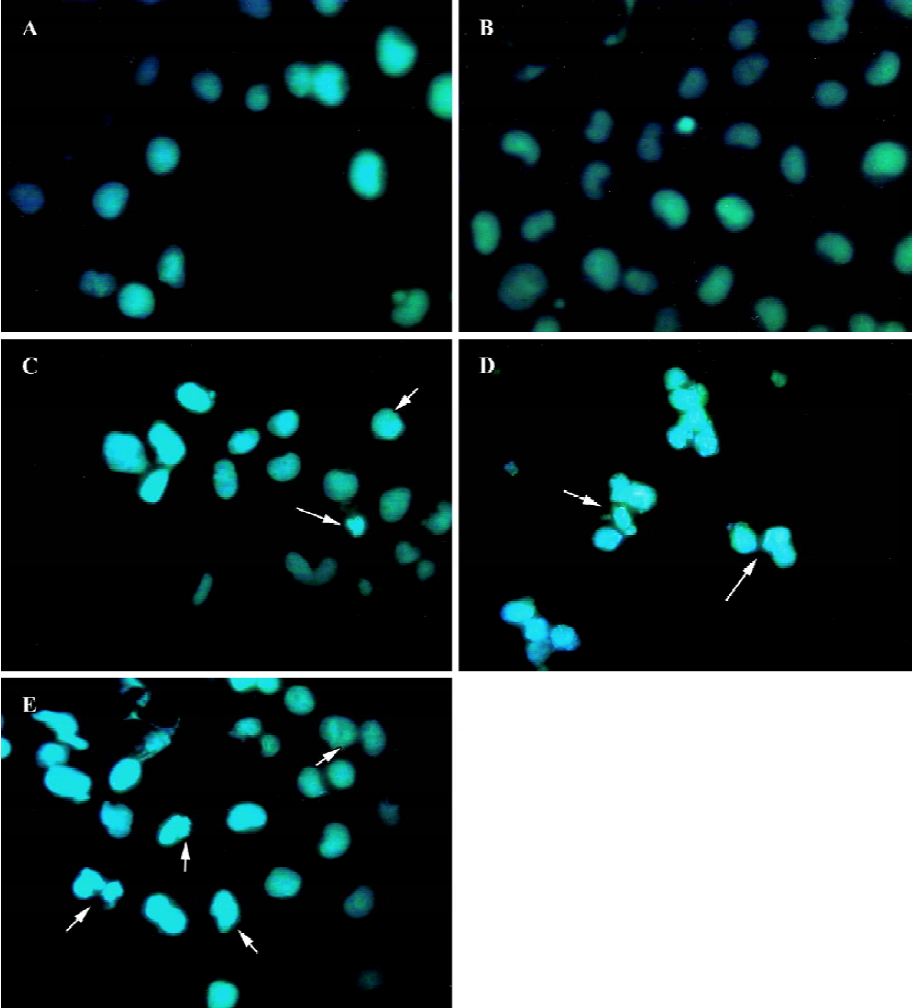
Morphology of fluorescence staining cells Hoechst 33258 is a specific fluorescent stain. A small quantity of Hoechst 33258 will be allowed to enter normal cells because of its natural characteristics. Cell membrane permeability will be enhanced during apoptosis, which makes more Hoechst 33258 enter apoptotic cells than natural cells to bind with base regions of DNA. At disposal of Hoechst 33258, the apoptotic body or fragments of nuclei were stained a deep blue color, normal cells a light blue color, and the delimitation between cytoplasm and nuclei of necrotic cells became unclear for organelle swelling and lysis through fluorescence microscope. From the photographs (Figure 2), apoptotic cells were nearly invisible from the untreated group and the empty pmU6 group, whereas the pmU6-RNAi groups (12 h, 24 h, and 48 h after transfection) had more apoptotic cells compared to the above mentioned groups. These results showed that the pmU6-RNAi plasmid efficiently induced apoptosis in CNE-2Z cells by expressing shRNA.
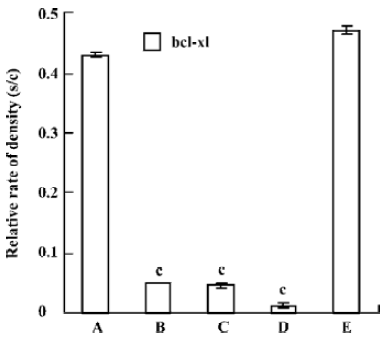
Quantitative RT-PCR The reduction of mRNA was in a time-dependent manner (Figure 3) apart from the untreated and the pmU6 treated group. The mRNA was reduced by 90% to 99% 12 to 48 h after transfection with pmU6-RNAi, respectively (Figure 4). The results indicate that the shRNA expressed by the pmU6-RNAi plasmid in cells could efficiently suppress bcl-xL gene expression.
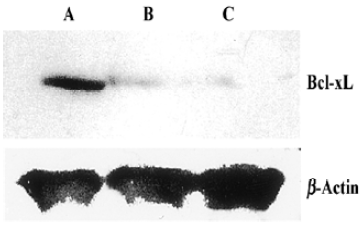
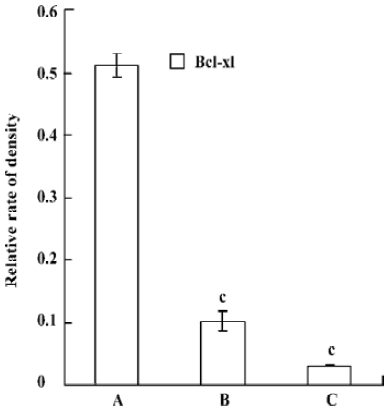
Western Blotting Western blot was used for the analysis of bcl-xL protein level. Compared with control, the pmU6-RNAi showed great capacities of inhibition at a protein level, and we also detected dramatic differences in the Bcl-xL protein levels between 24 h and 48 h after transfection, Bcl-xL protein was hardly measured after 48 h transfection (Figure 5, 6). This result suggested bcl-xL shRNA expressed by pmU6-RNAi could efficiently downregulate the protein level of Bcl-xL. Associated with the corresponding transfection time of bcl-xL mRNA levels and protein data, we found that bcl-xL shRNA could suppress bcl-xL gene expression, not only in transcript levels but also in protein levels.
Discussion
The expression level of some oncogenes such as bcl-2/bcl-xL, survivin, and caspase is significantly related to the prognosis of NPC. Particularly, overexpression of bcl-xL is widely considered as a poor prognosis following a tumor resection and might be associated with increased resistance to cancer chemotherapy[17,18]. Presently, radiotherapeutics and chemotherapeutics are the main therapies for this malignancy, and usually result in a favorable clinical response and a dramatic regression of NPC. But they have various kinds of unwanted side effects and locally advanced or metastatic NPC severely affect the prognosis in these patients and these therapies also fail to deliver a significant improvement in 5 year survival. It has been shown that specific suppression of over-expressed bcl-xL genes might result in an improvement in the therapy for this cancer. RNA interference, an useful tool for inhibiting the overexpression of specific oncogenes, was involved in the study of therapies for NPC in our study.
The Bcl-2 family of proteins play major role in regulating apoptosis and include both anti- and pro-apoptotic members[19]. The bcl-xL is an important anti-apoptotic member of the bcl-2 family and it has 44% homology to bcl-2. Its function is similar to bcl-2 in anti-apoptosis. Expression of this protein is significantly higher in NPC where it might play a pivotal role in tumor initiation, progression, and resistance to chemotherapy and radiotherapy[20,21]. In the present study, we constructed a plasmid expressing bcl-xL shRNA that could suppress bcl-xL gene expression in cells, and results indicated that bcl-xL shRNA could efficiently inhibit the expression of the target genes. According to the morphology of Hoechst 33258 staining cells, more apoptotic cells could be seen from the pmU6-RNAi transfected group than the empty plasmid group and the untreated group. RT-PCR analysis and Western-blot assay further indicated bcl-xL shRNA expressed by pmU6-RNAi had silenced the target gene and induced apoptosis in NPC cells.
Our results showed an obvious difference of time course between inducing of apoptotic cells by Hoechst 33258 analysis and decrease of bcl-xL mRNA by quantitative RT-PCR analysis (compare Figure 2 to Figure 3). The change at the mRNA level in pmU6-RNAi (12 h) transfected cells was obvious, whereas apoptotic cells were observed until 24 h after tranfection. This strongly suggested that down regulation of bcl-xL mRNA and protein, thereafter, by shRNA was the reason for apoptosis in NPC cells. The apoptosis occurred approximately 12 h after diminution of bcl-xL mRNA and finally the protein, to a definitive threshold.
Quantitative RT-PCR analysis of mRNA extracted from pmU6-RNAi transfected CNE-2Z cells was used to compare mRNA levels of bcl-xL with the corresponding protein data. From the analysis of RT-PCR, it showed that the bcl-xL mRNA levels between the empty plasmid group and the untreated group had no significant difference, and the results of quantitative RT-PCR analysis of bcl-xL revealed mRNA variations generally correlated with the Western blot data in our study. Another more interesting discovery was that pmU6-RNAi plasmid reduced bcl-xL mRNA levels by nearly 9.5-fold 12 h after transfection, almost equal to the decrease in the mRNA levels 24 h after transfection, despite significant and consistent changes were not always equal between mRNA and protein[22]. The tendency for variations between them was a common coincidence in our present study. For example, pmU6-RNAi treated for 24 h reduced mRNA levels by nearly 9.5-fold, and the reduction at the protein level was 8.5-fold. In another example, we detected a >9.9-fold decrease in the mRNA level by pmU6-RNAi (48 h) treated group, whereas the corresponding Western blots showed a 9.4-fold diminution in bcl-xL protein (compare Figure 4 to Figure 6). According to the above analysis, we presumed that there were similar changes between the protein level and the corresponding mRNA level of bcl-xL in pmU6-RNAi (12 h) treated group.
Based on the RNAi-mediated bcl-xL gene silencing in NPC cell line described above, we underlined the effects of bcl-xL shRNA to suppress bcl-xL gene expression. Taken together, these issues will have a profound influence on the use of RNAi technology, on a large scale, for functional genomics studies and therapy of cancer.
Footnote
Project supported by grants from the Natural Science Foundation of Guangdong Province (N
References
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-11.
- Sui GC, Christina SH, Affar EB, Gay FR, Shi YJ, Forrester WC, et al. DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002;99:515-20.
- Schuck S, Manninen A, Honsho M, Joachim F, Simons K. Generation of single and double knockdowns in polarized epithelial cells by retrovirus-mediated RNA interference. Proc Natl Acad Sci USA 2004;101:4912-7.
- Sunita G, Rebecca AS, James EE, Gregory JH, Vivek M. Inducible, reversible, and stable RNA interference in mammalian cells. Proc Natl Acad Sci USA 2004;101:1927-32.
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494-8.
- Brummelkamp HR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550-3.
- Monika P, Michael M, Mark I, Michal M, Yen C, Aaron K. Inhibition of herpes simplex virus 1 gene expression by designer zinc-finger transcription factors. Proc Natl Acad Sci USA 2003;100:1621-6.
- Zhang L, Yang N, Mohamed-Hadley A, Rubin SC, Coukos G. Vector-based RNAi, a novel tool for isoform-specific knock-down of VEGF and anti-angiogenesis gene therapy of cancer. Biochem Biophys Res Commun 2003;303:1169-78.
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, et al. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993;74:597-608.
- Altschul L, Stephen F, Thomas LM, Alejandro AS, Zhang JH, Zhang Z, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389-402.
- Xu F, Zhen YS. (-)-Epigallocatechin-3-gallate enhances anti-tumor effect of cytosine arabinoside on HL-60 cells. Acta Pharmacol Sin 2003;24:163-8.
- Zheng XL, Sun HX, Liu XL, Chen YX, Qian BC. Astilbic acid induced COLO 205 cell apoptosis by regulating Bcl-2 and Bax expression and activating caspase-3. Acta Pharmacol Sin 2004;25:1090-5.
- Wan YF, Guo XQ, Wang ZH, Ying K, Yao MH. Effects of paclitaxel on proliferation and apoptosis in human acute myeloid leukemia HL-60 cells. Acta Pharmacol Sin 2004;25:378-84.
- Liu LH, Wang XK, Hu YD, Kang JL, Wang LL, Li S. Effects of a fatty acid synthase inhibitor on adipocyte differentiation of mouse 3T3-L1 cells. Acta Pharmacol Sin 2004;25:1052-7.
- Li XH, Li JJ, Zhang HW, Sun P, Zhang YL, Cai SH, et al. Nimesulide inhibits tumor growth in mice implanted hepatoma: overexpression of Bax over Bcl-21. Acta Pharmacol Sin 2003;24:1045-50.
- Liu RY, Luo HL, Feng HL, Peng JL, Cai TY, Huang WL, et al. A method to detect gene transfection efficacy by flow cytometry. Chin J Cancer 2002;21:267-71.
- He GR, Zhou KY, Cai KR, Liang T, He CW, Zhang YF. Apoptotic effect of bcl-xL antisense oligodeoxynucleotide mediated by lipofectin on cell strain CNE-2Z. Chin J Cancer 2003;22:11-5.
- Min L, Zhou KY, Liang T, Zhang YF. Inhibitory effects of bcl-xL antisense oligodeoxynucleotides on growth of human nasopharyngeal carcinoma in nude mice. Chin J Oncol 2004;26:14-6.
- Yang SP, Song ST, Song HF. Advancements of antisense oligonucleotides in treatment of breast cancer. Acta Pharmacol Sin 2003;24:289-95.
- He GR, Zhou KY, Zhang YF, Liang T, Min L. Effect of lipofectin-mediated bcl-xL antisense oligodeoxynucleotide on CNE-2Z cells. J Guangdong Med Coll 2003;21:1-4.
- Reed JC. Prevention of apoptosis as a mechanism of drug resistance. Hem Onc Clinics North Am 1995;9:451-73.
- Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 2004;101:1892-7.