
Ca2+ participates in α1B-adrenoceptor-mediated cAMP response in HEK293 cells1
Introduction
It is well known that the classic signaling pathway of α1-adrenoceptor (AR) is to couple with the Gq/11 protein and then stimulate phosphatidylinositol turnover[1,2]. However, it has been recently discovered that α1-AR can also stimulate adenosine 3':5'-cyclic monophosphate (cAMP) accumulation in several cell lines, tissues and organs[3–12]. Our previous study showed that each of the three α1-AR subtypes was able to mediate cAMP generation in human embryonic kidney 293 (HEK293) cells. As for the signal transduction pathway by which α1-AR mediates cAMP production, several studies have shown that cAMP production is secondary to the α1-AR-induced phospholipase C (PLC)-phosphokinase C (PKC) stimulation[10–12], which cross-talked with AC[13]. Horie et al[14] found that in a Chinese hamster ovary (CHO) cell line transfected with α1B-AR, none of the PKC inhibitor, the Ca2+ ionophore, or the pertussis toxin (PTX) was able to inhibit NE-induced cAMP accumulation, whereas anti-Gsα antiserum inhibited the response, which suggests that α1B-AR activates AC and increases intracellular cAMP by directly coupling with Gsα. In fact the signal transduction pathway involved in α1B-AR-mediated cAMP generation remains unclear. Therefore, we transfected HEK293 cells (human embryonic kidney 293 cell line, which does not express any other adrenoceptors except native β2-AR) with full-length cDNA encoding α1B-AR and selected for subcloning cell lines stably expressing α1B-AR. This was a good model to investigate the regulating effects of the PLC-PKC pathway, the tyrosine kinase pathway, the Ca2+ signal system, and G proteins on cAMP production.
Materials and methods
Norepinephrine (NE), phenylephrine (PE), methoxamine (ME), prazosin (PRZ), propranolol (Prop), Triton X-100, cAMP, 3-isobutyl-1-methyl-xanthine (IBMX), pyruvic acid, hygromycin B, cyclopiazonic acid (CPA), phorbol 12-myristate 13-acetate (PMA), genistein, tyrphostin A25, egtazic acid (EGTA), nifedipine (Nif), CdCl2, PTX, and cholera toxin (CTX) were from Sigma Chemical Co (St Louis, USA); BAPTA/AM, Ro-31-8220, calphostin C, and W-7 were products of Calbiochem-Novabiochem International (San Diego, USA). BE2254 {[2-β(4-hydroxyphenyl)-ethylaminomethyl]-tetralone} was from Beiersdorf Co (Ham-burg, Germany); [3H]adenine was from Amersham Biosciences (Piscataway, United States); 2,5-diphenyl-oxazole (PPO) was bought from Farco-Pharma (Koln, Germany); fetal bovine serum (FBS) and Dulbecco’s Modified Eagle’s Medium (DMEM) were from Hyclone China (Beijing, China); HEK293 (human embryonic kidney 293) cells and full-length cDNA of hamster α1B-AR (pREP4) were kindly provided by Prof Kenneth P MINNEMAN (Emory Univer-sity, USA).
Transfection of HEK293 cells with cDNA encoding α1B-AR using the calcium phosphate precipitation method HEK293 cells were cultured in DMEM containing 10% FBS at 5% CO2 at 37 ºC. The cells were transfected with pREP4/α1B-AR by calcium phosphate precipitation at 70% confluence and selected with hygromycin B (0.05 g/L). Three days later the cells were diluted and planted in a 96-well dish, with 0-5 cells in each well. After 2 to 3 cloning sessions, a cell line stably expressing α1B-AR was obtained. The cells were continuously cultured and kept in DMEM containing selective antibiotics.
Determination of α1B-AR expression by a saturation radioligand binding assay The cells were grown in 75-mL flasks and harvested in PBS. After centrifugation at 3000×g, 4ºC for 10 min then at 21 000×g, 4ºC, for 20 min, the pellet was resuspended with 30 mL PBS and kept on ice. The ligand-binding properties of the receptors were determined in a series of radioligand binding studies using the α1-AR antagonist radioligand [125I]BE2254, which was radioio-dinated to theoretical specific activity as described by Engel and Hoyer[19]. Saturation reactions (total volume 250 µL) containing 100 µL or 50 µL PBS with 1% bovine serum albumin (BSA), 50 μL of [125I] BE2254 at different concentrations (15 000-500 000), and 100 µL cell membranes. Nonspecific binding was determined in the presence of 50 µmol/L phentolamine. Reactions were allowed to proceed for 20 min at 37 ºC. Reactions were terminated by adding 7 mL ice-cold Tris-HCl buffer (pH 7.4) and the mixture was filtered onto glass fiber filters. Filters were washed twice with 7 mL ice-cold Tris-HCl buffer and then dried. Bound radioactivity was measured using an auto-gamma counter. Binding data were analyzed by using nonlinear regression and Scatchard analysis (GraphPad Prizm Software) on a computer and thus the dissociation constant (KD) between the receptor and antagonist and maximal bound capacity (Bmax) could be obtained. Protein content was determined by using the Coomassie protein quantitation method.
cAMP determination in intact cells using ion-exchange chromatography HEK293 cells expressing α1B-AR were cultured in 24-well dishes at 37 ºC, 5% CO2, with 2.5×105 cells per mL medium. When the cells were fully confluent, the medium was changed, and [3H]adenine 18.5 MBq (0.5 mCi) was added into each well. After incorporation for 4 h at 37 ºC in 5% CO2, the medium was discarded and the cells were washed twice with pre-warmed Krebs’ solution. After addition of antagonists in 1 mL Krebs’ solution containing 200 µmol/L IBMX and incubation for 30 min, the cells were incubated for a further 20 min with different concentrations of agonists. The reaction was terminated by adding 100 µL of 77% trichloracetic acid, followed by a centrifugation at 3000×g, 4 ºC, for 20 min. Then 50 µL supernatant was transferred into 3 mL scintillation liquid to measure radioactivity as total activity (cpm). The remaining supernatant was applied to Dowex columns and aluminal columns. After being washed with distilled water, the aluminal columns were eluted with 2 mL Tris-HCl (pH 8.0), and the radioactivities of the eluates were measured as newly-produced cAMP. cAMP accumulation is equal to proportion of total radioactivity represented by newly-produced cAMP as a percentage. The formula is as follows:
Twenty-two is the volume constant.
Statistical analysis Results are expressed as mean±SD. To compare mean values between two groups, Student’s t-test was used; ANOVA was used for comparison among three or more groups. P<0.05 was considered statistically significant.
Results
Density of α1B-AR HEK293 cells transfected with α1B-AR were cultured under selective pressure from hygromycin B for several passages, thus we obtained a cell line stably expressing α1B-AR. The density of α1B-AR was 2004±138 pmol/g as determined by a radioligand binding assay (n=5, data not shown).
cAMP accumulation induced by stimulation of α1B-AR In blank HEK293 cells, NE (100 nmol/L–30 µmol/L), PE (100 nmol/L–300 µmol/L), or ME (1 µmol/L–1 mmol/L) did not cause cAMP accumulation in the presence of propranolol, an antagonist of β-AR (data not shown).
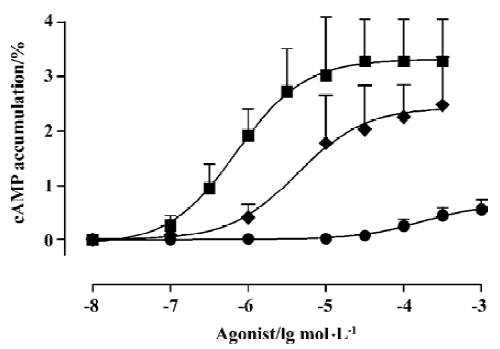
However, NE, PE, and ME all increased cAMP accumulation in HEK293 cells transfected with α1B-AR in a dose-dependent manner, with Rmax/pD2 of (3.32%± 0.34%)/(6.15±0.33) (n=7), (2.43%±0.46%)/(5.37±0.55) (n=8), and (0.66%±0.17%)/(3.79±0.39) (n=6), respectively (Figure 1), all of which were antagonized by prazosin (100 nmol/L) (Data not shown).
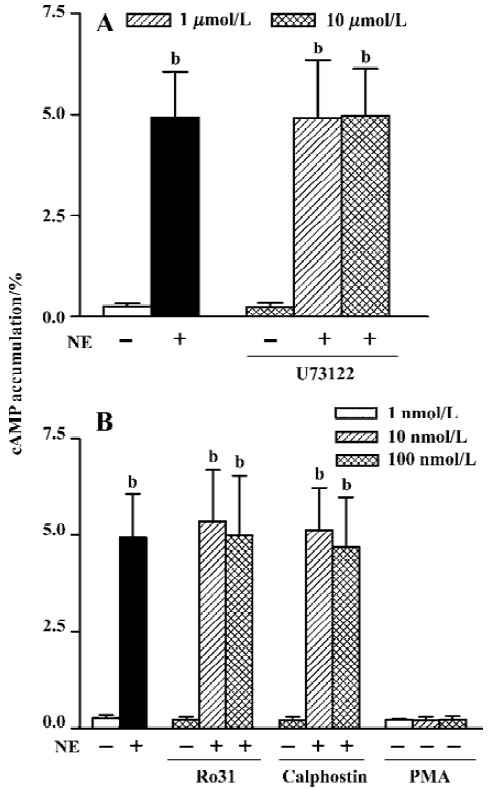
PLC-PKC pathway In the presence of propranolol (10 µmol/L) to block β2-AR in HEK293 cells, NE (10 µmol/L) increased cAMP accumulation from a basal level of 0.28%±0.07% to 4.93%±1.13% (n=11, P<0.01). The PLC inhibitor, U73122, had no effect on the NE-induced cAMP accumulation at 1 µmol/L (4.91%±1.43%, n=11) or 10 µmol/L (4.97%±1.15%, n=11, Fig 2A) for 1 h incubation. Neither U73122 itself (0.26%±0.09%, n=5) nor the vehicle alone (Me2SO, 0.1% v/v final, 0.26%±0.05%, n=3) affected the basal level of cAMP (0.28%±0.07%, n=11). Similarly, when PKC inhibitors were added, neither Ro31-8220 (5.37%±1.33%) and (4.99%±1.56%, n=8) nor calphostin C (5.14%±1.09%) and 4.70%±1.28%, n=8) at 10 nmol/L and 100 nmol/L affected NE-induced cAMP production (4.93%±1.13%, n=11, Figure 2B). The two inhibitors had no effect on the basal cAMP level (0.24%±0.07%, n=5, and 0.22%±0.09%, n=5, respectively, vs 0.28%±0.07%, n=11). To further clarify the role of PKC in this response, we examined whether PKC activator had any effect on cAMP synthesis. Without any AR agonist or antagonist, pretreatment with 1, 10, and 100 nmol/L PMA did not increase cAMP synthesis, the levels of which were (0.23%±0.03%, n=7), (0.22%±0.08%, n=7), and (0.24%±0.08%, n=7), respectively, showing no significant difference compared with the basal level (0.28%±0.07%, n=11).
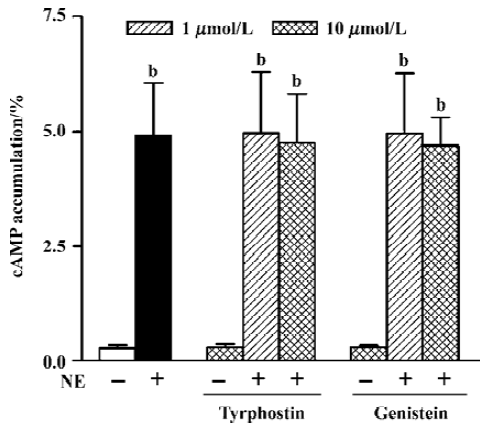
Tyrosine kinase signaling pathway The effects of two kinds of tyrosine kinase inhibitors, tyrphostin A25 and genistein, on NE-induced cAMP synthesis were examined. Tyrphostin A25 and genistein (0.30%±0.07% and 0.30%±0.04%, n=5, respectively) or the vehicle alone (Me2SO, 0.1% v/v final, 0.26%±0.03%, n=3) had no effect on the basal cAMP level (0.28%± 0.07%, n=11). After pretreating cells with tyrphostin A25 or genistein at 1 µmol/L and 10 µmol/L for 1 h, neither tyrphostin A25 (4.98%±1.33% and 4.75%±1.07% at the two concentrations, respectively, n=7) nor genistein (4.97%±1.30% and 4.69%±0.62%, respectively, n=7) influenced NE-induced cAMP synthesis (4.93%±1.13%, n=11 in the presence of propranolol 10 µmol/L) (Figure 3).
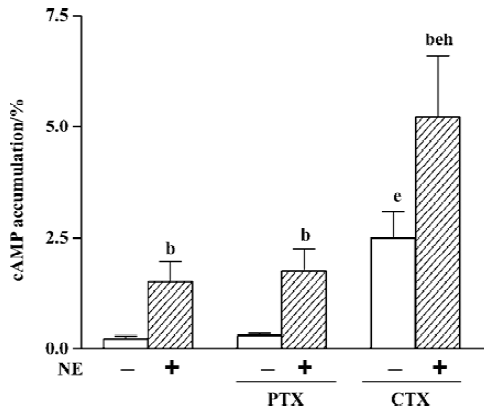
Regulation of cAMP response by G proteins After incubating cells with PTX 500 μg/L for 16 h, the effects of PTX on basal or NE-induced cAMP accumulation were examined. The results showed that PTX neither had any effect on basal cAMP (0.31%±0.07% vs 0.23%±0.07%, n=8) nor affected the NE-induced cAMP response (1.76%±0.50% vs 1.52%±0.44%, n=8, Figure 4). In order to examine whether α1-AR mediates cAMP response by directly coupling with the Gs protein, we compared cAMP accumulation upon stimulation of the Gs protein after incubation with 500 μg/L CTX for 3 h with NE-induced cAMP accumulation. The results showed that cAMP accumulation induced by CTX alone was significantly higher than that induced by 10 µmol/L NE (2.50%±0.60% vs 1.52%±0.44%, n=8, P<0.05), and when CTX and NE were used simultaneously, cAMP accumulation (5.24%±1.37%, n=8) was significantly higher than their combined effect when used alone (P<0.05, Figure 4).
Regulating effects of Ca2+ signaling system Because α1-AR can induce the release of the IP3-sensitive Ca2+ store, CPA, a Ca2+-ATPase inhibitor, was used to block Ca2+ being taken in sarcoplasmic reticulum again so as to increase [Ca2+]i and deplete the Ca2+ store. The results showed that CPA 10 µmol/L alone did not increase cAMP accumulation (0.20%±0.03% vs 0.22%±0.12%, n=8), neither did it have any effect on NE-induced cAMP production (1.63%±0.56%, n=8 vs 1.59%±0.47%, n=9, Figure 5A).
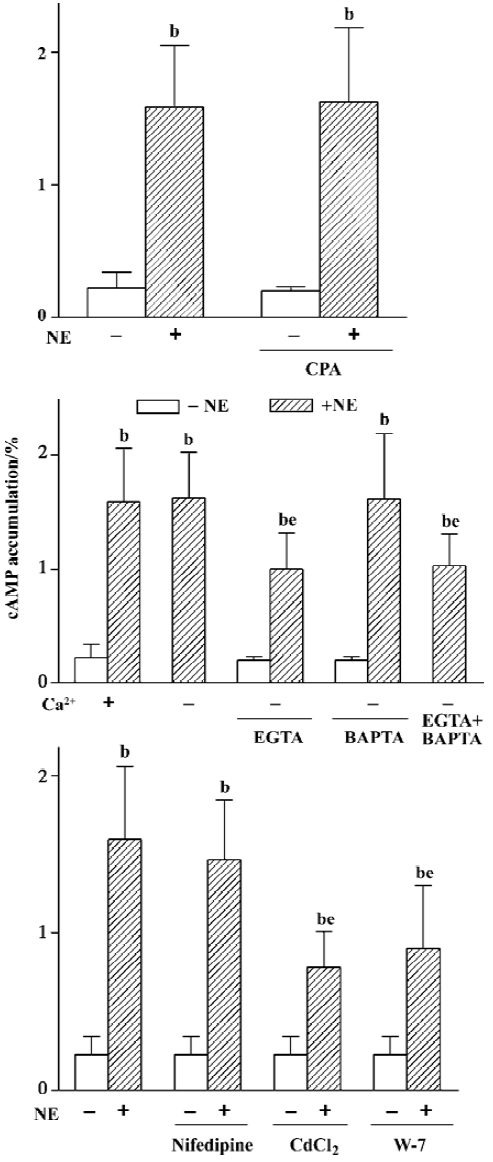
In addition, the influence of the Ca2+ influx on cAMP synthesis was studied. It was found that NE-induced cAMP accumulation under Ca2+-free condition was equal to that when Ca2+ was present (1.63%± 0.40% vs 1.59%±0.47%, n=9). However, when an extracellular Ca2+ chelator, EGTA (50 µmol/L), was added in Ca2+-free Krebs buffer to pretreat the cells for 1 h, the NE-induced cAMP synthesis was obviously reduced (1.00%±0.31%, n=9, P<0.05, Figure 5B), whereas 10 µmol/L intracellular Ca2+ chelator, BAPTA/AM, had no effect on the NE-induced cAMP response under Ca2+- free conditions (1.62%±0.58% vs 1.59±0.47%, n=9). There was no further inhibition on cAMP synthesis when BAPTA was used in combination with EGTA (1.03%±0.28% vs 1.00%±0.31%, n=9, Figure 5B).
To find out what type of Ca2+ channel admitted Ca2+, we examined the effects of two kinds of Ca2+ channel blockers on cAMP response and found that nifedipine 10 µmol/L (L-type Ca2+ channel blocker) did not affect the NE-induced cAMP synthesis (1.47%±0.38% vs 1.59%±0.47%,n=9), whereas CdCl2 1 mmol/L (nonselective Ca2+ channel blocker) obviously reduced NE-induced cAMP accumulation (from 1.59%±0.47% to 0.78%±0.23%, n=9, P<0.05, Figure 5C). Moreover, when pretreating cells with W-7, a calmodulin (CaM) inhibitor, NE-induced cAMP synthesis decreased markedly (0.90%± 0.40%, n=8, P<0.05, Figure 5C), and the extent of decrease was similar to that induced by EGTA or CdCl2 (P>0.05), while neither of them had any effect on the basal cAMP level.
Discussion
Several studies have demonstrated that α1B-AR mediates the cAMP response in HEK293 cells. However, the signaling pathway through which α1-AR mediates the cAMP response is unknown. Many studies have indicated that besides its classical signaling pathway, α1-AR can stimulate many other signal transduction pathways as well, such as the tyrosine kinase pathway[15-18], the phospholipase A2-arachidonic acid (PLA2-AA) signaling system[20], etc. Moreover, α1-AR also associates with other signaling pathways through its classical signal transduction pathway and consequently produces cross-talk. cAMP is a second messenger, a product of ATP catalyzed by adenylyl cyclase (AC), and a substrate of phosphodiesterase (PDE). There are at least 10 isozymes of AC. Besides the Gs and Gi proteins, many factors have been found to regulate their activities[21,22]. In the present study, we investigated the underlying mechanism involved in α1-AR-mediated cAMP synthesis using HEK293 cells transfected with α1B-AR.
PLC-PKC pathway α1-AR preferentially activates its classical signaling pathway. It stimulates PLC by coupling with the Gq/11 protein, then PLC hydrolyzes PIP2 to produce IP3 and DAG, which induces Ca2+ release and stimulates PKC, thereby producing biological effects. It has been reported that in some cell lines transfected with α1-AR subtypes, α1-AR first stimulates PI turnover to activate PKC, and then stimulates AC. This means that α1-AR-mediated cAMP response is secondary to the PLC-PKC pathway[10-12]. However, our results indicated that neither the PLC antagonist, U73122, nor the PKC antagonists, Ro31 and calphostin C, inhibited α1-AR-mediated cAMP synthesis, and the PKC activator, PMA, did not increase cAMP accumulation. Furthermore, there was no influence on cAMP synthesis when using the Ca2+-ATPase inhibitor, CPA, to block Ca2+ restoration and thus deplete the Ca2+ pool. All of the above suggests the taches lying in downstream of PLC in the classical pathway are not connected with α1-AR-mediated cAMP response.
Tyrosine kinase signal transduction pathway The tyrosine kinase signal transduction pathway relates closely to the classical signaling pathway of α1-AR. Some studies have shown that tyrosine kinase probably participates in α1-AR-mediated biological effects[15-18]. In our previous study, we found that the major functional α1-AR subtype of the vascular bed in rat hind legs was α1A-AR, and that tyrosine kinase antagonists could dose-dependently reduce the vascular contractive response to NE. Moreover, tyrosine kinase antagonists can inhibit α1-AR-mediated increase in intracellular Ca2+ in HEK293 cells[23], showing that α1-AR can activate tyrosine kinase. It is also known that tyrosine kinase can indirectly stimulate AC through the PLC-γ-IP3/DAG pathway, which results in intracellular Ca2+ mobilization and stimulation of PKC[22]. Since we have confirmed that α1-AR does not mediate cAMP synthesis through the PLC-PKC pathway in HEK293 cells, the possibility mentioned above can be excluded. Although it is known that tyrosine kinase can indirectly stimulate AC through the PLC-γ-IP3/DAG pathway[21], this would not happen in HEK293 cells because our study indicated that the PLC-PKC pathway was not involved in α1-AR-mediated cAMP accumula-tion. However, whether tyorsine kinase plays a role through other pathways or taches is unclear. In our present study, two kinds of tyrosine kinase inhibitors, different in structure and mechanism, exerted no influence on NE-induced cAMP synthesis, suggesting that tyrosine kinase is not involved in α1B-AR-mediated cAMP response in HEK293 cells.
Regulation of cAMP response by G proteins α1-AR is a typical G protein coupled receptor, whose classical signaling pathway is to couple with the Gq/11 protein. But it has been found that α1-AR also couples with Gs and Gi pro-teins[14,24]. Overexpressed α1-AR in the heart of transgenic mice can couple with the Gi protein and inhibit AC[24]. Horie et al[14] found that stimulation of transfected α1-AR directly activated Gs and increased cAMP accumulation. These phenomena occur when the density of α1-AR is much higher than normal. Some researchers[14,25] consider that receptor-G promiscuity happens when receptors are overexpressed, ie, receptors not only couple with their traditional G proteins, but also couple with irrelevant G proteins under normal conditions, and then produce new biological effects. This phenomenon is called receptor-G protein promiscuity, which would occur when receptors are overexpressed[14,25]. Since our experiments were performed under similar conditions, this leads to the following question: did promiscuity occur between α1-AR and other G proteins? Firstly, the results showed α1B-AR-mediated cAMP response was not affected by PTX, thereby excluding the possibility that Gi protein or Gβγ, which is sensitive to PTX[27] and activates ACII and IV[21,22,28], which is involved in the response. Secondly, cAMP accumulation induced by CTX alone was markedly higher than that induced by NE, and when CTX and NE worked together cAMP accumulation was far higher than the sum of cAMP accumulation when each of them worked alone. The response induced by NE 10 μmol/L almost reached a maximum, and since there is no receptor reserve in cloned HEK293 cells[25], if α1B-AR directly coupled with the Gs protein while the number of Gs were relatively insufficient or equally by the number of α1B-AR, cAMP accumulation induced by NE should equal that induced by CTX, and should not increase further when NE was added together with CTX. However, it did not occur. Thus there are two possibilities, one is that α1B-AR directly couples with the Gs protein but that the amount of Gs protein far exceeds what α1B-AR requires; another is that α1B-AR itself does not couple with the Gs protein, which is more likely, because in our study, stimulation of α1B-AR and the Gs protein at the same time induced a synergistic effect. In addition, the Gα, Gβγ subunit should not be ignored, because it has been shown to activate ACII and IV[21,22,28]. However, because Gβγ is PTX-sensitive[27], and PTX did not inhibit NE-induced cAMP sythesis in our study, we conclude that Gβγ does not participate in the response.
Regulating effects of Ca2+ signaling system α1-AR can induce the mobilization of intracellular Ca2+ as well as the influx of extracellular Ca2+. As the most ubiquitous and most active second messenger in cells, Ca2+ not only mediates many physiological effects directly, but also regulates many signaling pathways and molecules. It has been shown that the Ca2+-CaM complex can stimulate ACI, III, and VIII[21,22]. For this reason the effect of Ca2+ on α1B-AR-mediated cAMP response was investigated. Our results showed that the Ca2+-ATPase inhibitor, CPA, had no effect on NE-induced cAMP accumulation, indicating that mobilization of intracellular Ca2+ was not involved in the response. NE-induced cAMP accumulation was not reduced under Ca2+-free condition, but reduced after addition of the extracellular Ca2+ chelator, EGTA. We speculate that it is because even under Ca2+-free conditions, there inevitably existed a little Ca2+ in the buffer, which is enough to meet with the needs of the α1B-AR-mediated cAMP response. When extracellular Ca2+ is chelated by EGTA, Ca2+ cannot flow into cells, thereby reducing cAMP production. So cAMP production was reduced. All of the above shows that extracellular Ca2+ influx plays a role in the α1B-AR-mediated cAMP response. It is well known that extracellular Ca2+ enters cells mainly via voltage-dependent Ca2+ channels (VDCC) or voltage-independent Ca2+ channels, which includes Ca2+ store depletion-dependent Ca2+ channels (SDDCC), receptor-dependent Ca2+ channels, and so on. Then arises the new question of what kind of channel is involved in this response. It has been proving that there is no VDCC in HEK293 cells[29], and in our study, the L-type Ca2+ channel inhibitor nifedipine did not affect NE-induced cAMP sythesis. The nonselective Ca2+ channel inhibitor CdCl2 obviously reduced NE-induced cAMP accumu-lation, and the extent of reduction was the same as that with EGTA. If these facts are taken together with the fact that CPA-induced Ca2+ store depletion did not increase cAMP accumulation, it seems that it was via receptor-dependent Ca2+ channels that extracellular Ca2+ entered the cells. CaM inhibitor W-7 significantly reduced NE-induced cAMP accumulation, with the extent of reduction almost equal to that induced by EGTA and CdCl2, which indicates that it is Ca2+-CaM that activates AC. Intracellular Ca2+ chelator BAPTA can chelate Ca2+ released from the Ca2+ store as well as Ca2+ entering cells from the outside, so theoretically it should have an effect similar to EGTA. In fact, it neither inhibited NE-induced cAMP synthesis nor enhanced the inhibitory effect of EGTA. As for the reason why intracellular Ca2+ chelator BAPTA could not inhibit NE-induced cAMP synthesis, we suppose either that the penetrability of BAPTA/AM into HEK293 cells was rather weak, or that the activity of esterase in HEK293 cells is so low that BAPTA/AM cannot be degraded to active BAPTA. Furthermore, our previous study showed that [Ca2+]i increased to 4-5 times the basal level 8 s after activation of α1-AR. Active BAPTA was perhaps not enough to chelate all intracellular Ca2+ completely and rapidly, so BAPTA/AM had little effect. However, the exact mechanism by which the system functions remains to be studied. In summary, the results suggest that stimulation of α1B-AR triggers the receptor-dependent Ca2+ channel via an unknown pathway and lets extracellular Ca2+ in, then Ca2+ links CaM into a Ca2+-CaM complex, which activates AC to increase cAMP synthesis. However none of EGTA, CdCl2, and W-7 are able to completely inhibit NE-induced cAMP accumulation, suggesting that the Ca2+ influx is only partly involved in the response.
In conclusion, by coupling with a PTX-insensitive G protein, α1B-AR promotes Ca2+ influx via receptor-dependent Ca2+ channels, then Ca2+ links to CaM to form a Ca2+-CaM complex, which stimulates adenylyl cyclase (AC) and thereby increases the cAMP production in HEK293 cell lines. But Ca2+ only partly contributes to α1B-AR-mediated cAMP accumulation. The other mechanisms remain to be investigated.
Footnote
Project supported by the National Natural Science Foundation of China (N
References
- Williams S, Meij JT, Panagia V. Membrane phospholipids and adrenergic receptor function. Mol Cell Biochem 1995;149/150:217-21.
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, et al. International union of pharmacology nomenclature. Pharmacol Rev 1994;46:121-36.
- Morgan NG, Blackmore PF, Exton JH. Age-related changes in the control of hepatic cyclic AMP levels by α1- and β2-adrenergic receptors in male rats. J Biol Chem 1983;258:5103-9.
- Morgan NG, Waynick LE, Exton JH. Characterization of the α1-adrenergic control of hepatic cAMP in male rats. Eur J Pharmacol 1983;96:1-10.
- Chan TM, Exton JH. α1-Adrenergic-mediated accumulation of adenosine 3´:5´-monophosphate in calcium depleted hepatocytes. J Biol Chem 1977;252:8645-51.
- Blair JB, James ME, Foster JL. Adrenergic control of glucose output and adenosine 3´:5´ monophosphate levels in hepatocytes from juvenile and adult rats. J Biol Chem 1979;254:7579-84.
- Johnson RD, Minneman KP. Differentiation of α1-adrenergic receptors linked to phosphatidylinositol turnover and cyclic AMP accumulation in rat brain. Mol Pharmacol 1986;31:239-46.
- Perkins JP, Moore MM. Characterization of the adrenergic receptors mediating a rise in cyclic 3´,5´-adenosine monophosphate in rat cerebral cortex. J Pharmacol Exp Ther 1973;185:371-8.
- Robinson JP, Kendall DA. Niguldipine discriminates between α1-adrenoceptor-mediated second messenger responses in rat cerebral cortex slices. Br J Pharmacol 1990;100:3-4.
- Schwinn DA, Page SO, Middleton JP, Lorenz W, Liggett SB, Yamamoto K, et al. The α1C-adrenergic receptor: characterization of signal transduction pathways and mammalian tissue heterogeneity. Mol Pharmacol 1991;40:619-26.
- Perez DM, DeYoung MB, Graham RM. Coupling of expressed α1B- and α1D-adrenergic receptors to multiple signaling pathways is both G protein and cell type specific. Mol Pharmacol 1993;44:784-95.
- Cotecchia S, Kobilka BK, Daniel KW, Nolan RD, Lapetina EY, Caron MG, et al. Multiple second messenger pathways of α1-adrenergic receptor subtypes expressed in eukaryotic cells. J Biol Chem 1990;265:63-9.
- Yoshimasa T, Sibley DR, Bouvier M, Lefkowitz JR, Caron MG. Cross-talk between cellular signalling pathways suggested by phorbol-ester-induced adenylate cyclase phosphorylation. Nature (Lond) 1987;327:67-70.
- Horie K, Itoh H, Tsujimoto G. Hamster α1B-adrenergic receptor directly activates Gs in the transfected Chinese hamster ovary cells. Mol Pharmacol 1995;48:394-400.
- Filipeanu CM, Brailoiu E, Huhurez G, Slatineanu S, Baltatu O, Branisteanu DD. Multiple effects of tyrosine kinase inhibitors on vascular smooth muscle contraction. Eur J Pharmacol 1995;281:29-35.
- Abebe W, Edwards JD, Agrawal DK. G-proteins in rat blood vessels-II: assessment of functional involvement. Gen Pharmacol 1995;26:75-83.
- Thorburn J, Thorburn A. The tyrosine kinase inhibitor, genistein, prevents α-adrenergic-induced cardiac muscle cell hypertrophy by inhibiting activation of the Ras-MAP kinase signaling pathway. Biochem Biophys Res Commun 1994;202:1586-91.
- Slupsky JR. Raf-dependent signaling pathways in cell growth and differentiation. Cell Growth Oncogenes 1998; 75–95.
- Engel G, Hoyer D. 125I-BE2254, a new high affinity radioligand for alpha1-adrenoceptors. Eur J Pharmacol 1981;73:221-4.
- Weiss BL, Insel PA. Intracellular Ca++ and protein kinase C interact to regulate alpha-1-adrenergic and bradykinin receptor stimulated phospholipase A2 activation in Madin-Darby carby canine kidney cell. J Biol Chem 1991;266:2126-33.
- Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol 1996;36:461-80.
- Dolphin AC. G-proteins. Textbook of receptor pharmacology. Boca Raton (USA): CRC Press, Inc; 1996. page 194–5.
- Zhu WZ, Gao BB, Li HW, Zhang YY, Han QD. Differential activation of adenylyl cyclase by protein kinase C isoenzymes. Acta Pharmacol Sin 1999;20:1025-30.
- Shahab A. Transgenic mice with cardiac overexpression of α1B-adrenergic receptors. J Biol Chem 1997;272:21253-9.
- Zhu X, Gilbert S, Birnbaumer M, Birnbaumer L. Dual signaling potential is common among Gs-coupled receptors and dependent on receptor density. Mol Pharmacol 1994;46:460-9.
- Minneman KP, Theroux TL, Hollinger S, Han C, Esbenshade TA. Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol Pharmacol 1994;46:929-36.
- Exton JH. Phosphoinositide phospholipase and G proteins in hormone action. Annu Rev Physiol 1994;56:349-69.
- Taussig R, Tang WJ, Hepler JR, Gilman AG. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J Biol Chem 1994;269:6093-100.
- Tao L, Guan YY, He H, Han C, Zhang YY, Sun JJ. Comparison of the Ca2+ movement by activation of alpha 1-adrenoceptor subtypes in HEK293 cells. Life Sci 1997;61:2127-36.