
Adenovirus-mediated expression of Tob1 sensitizes breast cancer cells to ionizing radiation1
Introduction
Although many improvements of therapeutic techniques, as well as new therapeutic methods, have been introduced into cancer treatment, radiotherapy by ionizing radiation continues to be the frontline treatment for many types of cancers, including breast cancer[1]. Ionizing radiation is an effective DNA-damaging agent, producing a range of lesions in cellular DNA, including base damages, single-strand breaks, double-strand breaks, DNA–DNA, and DNA–protein cross-links. Among them, DNA double-strand breaks are the most important for cell death by ionizing radiation. It is known that 2 major pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), are involved in Double Strand Break repair[5]. The proteins that are known to participate in such DNA repair include DNA-dependent protein kinases (DNA-PK), Ku70 and Ku80, and X-ray-sensitive complementation group 4 (XRCC4), and DNA ligase IV[6,7].
Tob1, a member of the Transducing Molecule of ErbB2/B-cell Translocation G gene (Tob/BTG) family, is an antiproliferative protein. Since the exogenous overexpression of the Tob1 protein induces G1 arrest via the suppression of cyclin D1, a major player in the G1 checkpoint of cell cycle progression[8,9]. Increasing evidence suggests that Tob1 may be a new tumor suppressor gene for the following reasons: (i) mice lacking the Tob1 gene rapidly develop tumors in lungs, liver, and lymph nodes[10]; (ii) the down-regulation of Tob1 expression is often observed in human cancers[11]; and (iii) the inactivation of Tob contributes to the progression of papillary carcinoma of the thyroid[12], but the increased expression of Tob1 protein suppresses tumor cell growth and cell cycle arrest[8,9,13]. The degradation of the Tob1 protein is through the ubiquitin–proteasome pathway by the Skip1/Cull/F-box protein-Skp2 ubiquitin ligase[14,15].
Enhancing the antitumor effects by combining ionizing radiation with other agents, including gene therapy, often allows lower doses to be used, thereby minimizing side-effects. Tob1 is considered to play a role in mediating radiosensitivity for the following reasons: (i) Tob1 inhibits the expression and promoter activity of cyclin D1. Cyclin D1 is most often overexpressed or amplified in breast cancer. Its high expression results in a decrease in radiosensitivity[16]; (ii) the activation of Extracellular Signal Regulated Kinases(ERK)1 and 2 induces Tob phosphorylation[17], while the downregulation of ERK2 is essential for the inhibition of radiation-induced cell death[18]; and (iii) Tob1 inhibits estrogen receptor-mediated transcriptional activation[19]. It is known that the estrogen receptor plays a major role in breast cancer development and mediates radiosensitivity[20,21]. To test our hypothesis, we employed a system with the adenovirus-mediated expression of Tob1 and found that the enhanced expression of Tob1 by the adenovirus-mediated expression of Tob1 increases the radiosensitivity of breast cancer cells via increasing apoptosis and repressing DNA repair by mediating the expression of pro-apoptotic protein Bax and several DNA Double Strand Break repair proteins.
Materials and methods
Cell culture and irradiation Invasive human breast cancer MDA-MB-231 cells were originally purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum, 100 unit/mL penicillin, 100 µg/mL streptomycin, and a mixture of non-essential amino acids (Sigma–Aldrich, St Louis, MO, USA) at 37 ℃ in an atmosphere of 5% CO2. Irradiation was performed with γ-radiation (JL Shepherd Mark I Radiator,) with a 137Cs source emitting at a fixed dose rate of 3.5 Gy/min.
Generation of recombinant adenoviruses The E1-deleted adenovirus-β-gal (Ad-β-gal) was obtained from Introgen Therapeutics (Houston, TX, USA). A recombinant adenovirus (pAd/CMV/V5-DEST; Invitrogen, USA) containing a 1038-base pair DNA fragment encoding the complete amino acid sequence of human Tob1 (Ad5-Tob1) between the CMV promoter and the polyadenylation signal (TK pA) was prepared as outlined in Figure 1. These adenoviral vectors were propagated in 293 human embryonal kidney cells (Invitrogen, USA) using the Stratagene MBS mammalian transfection kit (La Jolla, CA, USA) with a modified calcium phosphate transfection protocol. The transfected cells were incubated at 37 ℃ for 7 d, then harvested and subjected to freezing (liquid nitrogen)/thawing (in a 37 ℃ water bath) for 4 cycles. The cell lysates were centrifuged at 12 000×g for 10 min at 4 ℃, and the supernatant (primary virus stock) was transferred to a fresh screw-cap mini-centrifuge tube and stored at –80 ℃. Recombinant adenoviruses were further amplified using the same procedure; the cell lysates were centrifuged on cesium chloride step gradients at 60 000×g for 2 h at 4 ℃ to separate the viruses from defective particles and empty capsids. Recovered virus bands were dialyzed 3 times against phosphate-buffered saline (PBS). The viruses were aliquoted in a buffer containing 10 mmol/L Tris-HCl (pH 7.4), 10 mmol/L MgCl2, and 10% v/v glycerol and stored at -80 ℃. Under these conditions, there was no precipitation of virus particles or loss of virus infectivity due to inactivation or aggregation. To control the biological effect of the virus per se, the vector, Ad5-CMV-Null, expressing no transgene (Ad5), was constructed in a similar method, but without the subcloned gene sequences.
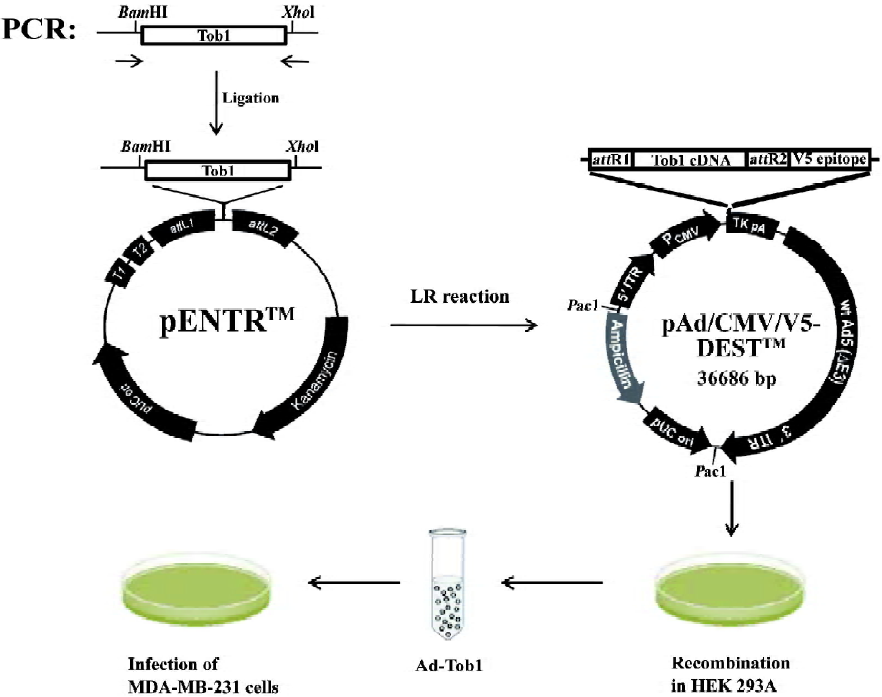
For the adenovirus infection, 2.5×104 cells in each well of a sex-well dish were infected with the appropriate amount of replication-defective adenoviruses with Ad-β-gal and incubated with gentle shaking for 2 h at 37 ℃. X-gal (1 mg/mL) was used to stain the cells for β-gal and the cells were sustained overnight. The cells were then fixed in 10% formalin, washed in PBS, and kept in PBS at 4 ℃. Blue-stained cells were considered to be infected with Ad-β-gal. Afterwards, fresh growth medium was added to each dish. To monitor the Tob1 expression, the infected cells were further incubated at 37 ℃ for various time-points and the Tob1 protein was then determined by Western blot assay. To examine radiosensitivity, the infected cells were incubated at 37 ℃ for 48 h before γ-ray irradiation; a colony formation assay was then performed.
Western blot assay The protein expression was detected by Western blot assay, as previously described[22]. The cells were harvested by trypsin and centrifugation. The cells were washed twice with ice-cold 1×PBS and lysed with a protein lysis buffer containing Tris-HCl [50 mmol/L (pH 7.4)], NP-40 (1%), Na-deoxycholate (0.25%), NaCl (150 mmol/L), EDTA (1 mmol/L), phenylmethylsulphonyl fluoride (PMSF) (1 mmol/L), Na3VO4 (1 mmol/L), NaF (1 mmol/L), and an inhibitor cocktail (Sigma–Aldrich, USA). After centrifuga-tion, the supernatant was transferred into new tubes. The protein concentration was determined by the Bio-Rad protein assay (Invitro-gen, USA). 50 µg total whole cell lysates were separated on SDS–PAGE and electroblotted onto nitrocellulose membranes (Millipore, Billerica, MA, USA). The membranes were then incubated in blocking solution of 5% non-fat milk in TBS-T (20 mmol/L Tris-HCl, 150 mmol/L NaCl, and 0.1% Tween-20), followed by overnight incubation with the appropriate primary antibodies. After completely washing with TBS-T and incubating with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h, immunocom-plexes were developed with an enhanced horseradish peroxidase/luminol chemiluminescence reagent (Sigma–Aldrich, USA) according to the manufacturer’s instruction. Primary antibodies included a mouse monoclonal anti-Tob1 antibody (4B1, 1:500 dilution) and a rabbit polyclonal anti-Ku80 antibody (557-570, 1:500 dilution), which were purchased from Sigma–Aldrich (USA). A mouse monoclonal anti-Ku70 antibody (A-9, 1:1,000 dilution), a mouse monoclonal anti-DNA–PK antibody (G-4, 1:1,000 dilution), a goat polyclonal anti-XRCC4 antibody (D-18, 1:500 dilution), an antigoat polyclonal antibody, and a goat polyclonal anti-actin antibody (I-19, 1:2,000 dilution) were obtained from Santa Cruz Biotechnology (USA).
Clonogenic assay Cell survival was evaluated by clono-genic assay as detailed in a previous study[23]. Briefly, the cells were trypsinized immediately after irradiation and counted. Known numbers were subcultured in 100 mm culture dishes in 2 sets of triplicates for each dose of radiation; sufficient numbers were seeded to ensure that about 50–100 macroscopic colonies would appear in each plate of untreated and uninfected control cells at the end of 15 d. The colonies were then fixed, stained, and counted. Surviving fractions were normalized by the plating efficiency of unirradiated controls (40%–60% for MDA-MB-231). a and b (constants determined by the linear quadratic model where S=e–[aD+bD2] and S is the fraction of cells surviving a dose [D]) parameters, as well as D0 (defined by slope of the terminal exponential region of the 2-component [single-hit multitarget model] survival curve where slope=1/D0), Dq (quasithreshold dose), and n (extrapolation number) values were calculated for com-parison.
DNA fragmentation gel electrophoresis Apoptosis induction was determined by a DNA fragmentation gel electrophoresis, as described in a previous study[23]. The cells were collected by trypsin and centrifuged for 5 min at 13 000×g. The cells were washed with 1×PBS and centrifuged again. The cell pellets were then lysated with 200 µL lysis buffer [10 mmol/L Tris-HCl (pH 7.6), 100 mmol/L EDTA, and 20 mmol/L NaCl] and centrifuged. The supernatant was transferred into new tubes. 20 µL SDS plus 200 µL RNase A (10 mg/mL, Sigma–Aldrich, USA) was added to each tube. After 2 h incubation at 56 ℃, 30 µL proteinase K (50 mg/mL, Sigma–Aldrich, USA) was added to each tube. The cell lysates were incubated at 37 ℃ for another 2 h. The DNA was finally precipitated with the addition of 10 µL of 10 mol/L potassium acetate and 1 mL 100% ethanol at –80 ℃ for 30 min. The extracted DNA samples were centrifuged and washed with 70% ethanol. Pure DNA were finally loaded and run on a 1% agarose gel at 80 V in running buffer (89 mmol/L Tris-acetate, 2 mmol/L Na2EDTA, and 89 mmol/L boric acid), stained with ethidium bromide and photographed. The samples were run in tandem with a DNA molecular weight ladder (Invitrogen, USA) providing molecular size markers of 0.5 to 12 kilobase pairs. Gel photographs were evaluated for typical ladder patterns of low molecular weight DNA fragments in multiples of 180–200 base pairs, a hallmark of apoptosis.
Determination of apoptosis by terminal deoxynucleotidyl transferase-mediated nick end labeling assay Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) was performed using the APO-BRDUTM kit (Phar-Mingen, San Diego, CA, USA) to quantify the induction of apoptosis following the manufacturer’s instructions. Briefly, the cells were fixed in 1% (w/v) paraformaldehyde in ice-cold PBS and incubated on ice for 15 min. Then the cells were washed twice with PBS and stored in 70% (v/v) ethanol overnight. About 1×106 cells/treatment in duplicate, along with positive and negative controls, were counted, pelletized, washed twice with wash buffer, and subjected to labeling reaction using terminal deoxynucleotidyl transferase overnight at room temperature. At the end of the reaction, the cells were rinsed twice before treatment with fluorescein-labeled anti-BrdU antibody solution in the dark for 30 min. The cells were stained with propidium iodide/RNase solution for 30 min in the dark and analyzed by flow cytometry (Epics XL-MCL, Beckman Coulter, Miami, FL, USA).
Host cell reactivation assay Ten thousand cells were plated in each well of 6-well plates. The cells were infected with Ad5-Tob1 or Ad5 at a multiplicity of infection (MOI) of 50 or 100 followed by 48 h incubation. The cells were then infected with Ad-beta-gal (1×103 vp/cell) that had been irradiated with 0–4000 Gy of γ-ray irradiation and incubated for an additional 24 h. This dose of 4000 Gy was necessitated by the small genome size of the adenoviral vector compared with a mammalian cell. The calculations indicated that this dose would produce about 1–2 DSBs/vector particles. The cells were then stained with X-gal following the procedure described above and fixed in 10% formalin. beta-gal-positive (blue) cells were scored under high power (×40) of a light microscope. The data are presented as the percentage of the control.
Statistics All experiments were repeated at least 3 times. The results are expressed as the mean±SEM. Statistical analysis was performed with 2-tailed Student’s t-test when 2 treatment regimens were compared. Probability values of P<0.05 were considered statistically significant.
Results
Adenovirus-mediated Tob1 gene expression increases sensitivity of MDA-MB-231 cells to ionizing radiation A significant infection of MDA-MB-231 cells with adenoviral vectors was confirmed by β-gal staining (data not shown) and the Tob1 protein expression were monitored by Western blot assay (Figure 2A). A time-dependent increase of Tob1 protein expression was obtained following the infection of Ad5-Tob1 at a MOI of 100 plaque-forming units/cell, starting at 24 h and reaching the maximum at 96 h, while till higher than uninfected control cells at 144 h. Control Ad5 infection did not affect Tob1 expression (data not shown). Greater than 95% transduction efficiency was detected in these cells (data not shown).
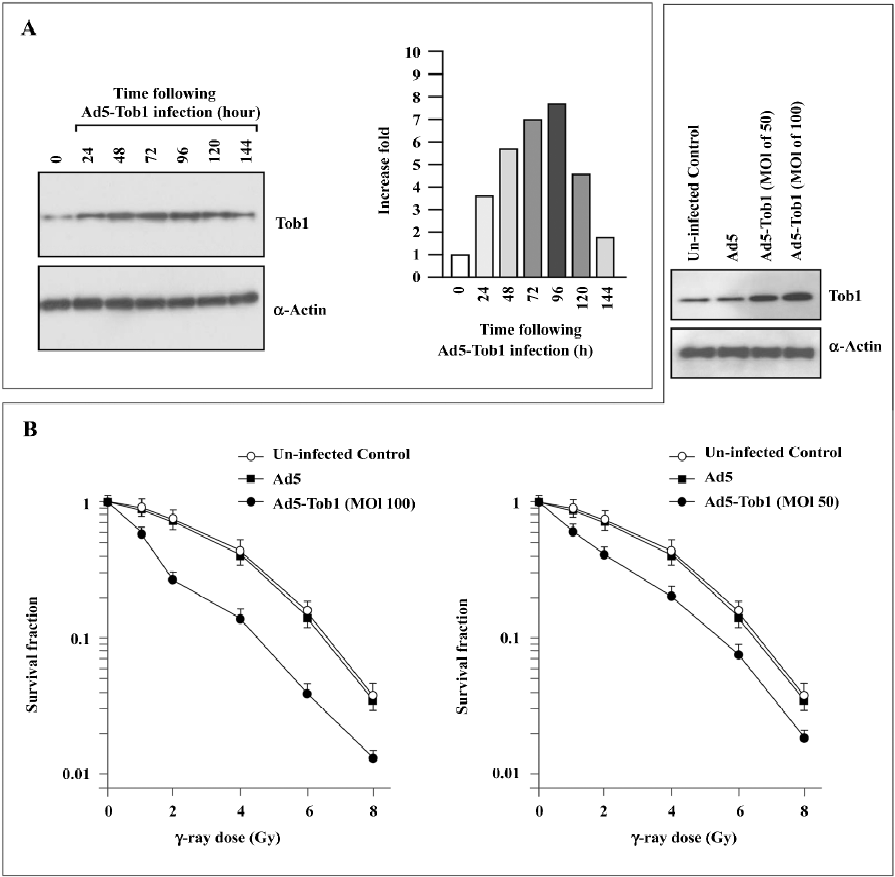
As illustrated in Figure 2B, MDA-MB-231 showed a typical clonogenic survival curve with a shoulder signifying cellular repair ability. The infection of the MDA-MB-231 cells with Ad5-Tob1 significantly increased cell susceptibility to γ-irradiation in an Ad5-Tob1 dose-dependent fashion. For example, doses of 2, 4, and 6 Gy of γ-radiation alone killed about 28%, 56%, and 85% of cells, respectively, while Ad5-Tob1 at the MOI of 100 enhanced cell death to 73% (P<0.01), 87% (P<0.01), and 97% (P<0.05) at 2, 4, and 6 Gy of γ-radiation, respectively. However, Ad5 (as a negative control) did not show any significant effects on radiosensitivity (Figure 2B). Survival curves were analyzed using the single-hit multitarget and linear quadratic models of cell survival. a and b (constants determined by the linear quadratic model where S=e–[aD+bD2] and S is the fraction of cells surviving a dose [D]) parameters as well as D0 (defined by slope of the terminal exponential region of the 2-component [single-hit multitarget model] survival curve where slope=1/D0), Dq (quasithreshold dose), and n (extrapo-lation number) values are summarized in Table 1. There were statistically significant differences in these values between the cells infected with Ad5 and the cells infected with AD5-Tob1 (P<0.01). Both Ad5-Tob1 and Ad5 at the MOI less than 100 did not produce any significant toxic effects in MDA-MB-231 cells (data not shown). These results suggested that the radiosensitizing effect of Ad5-Tob1 was mediated by the increased expression of Tob1 and was not due to a non-specific effect of the vector.
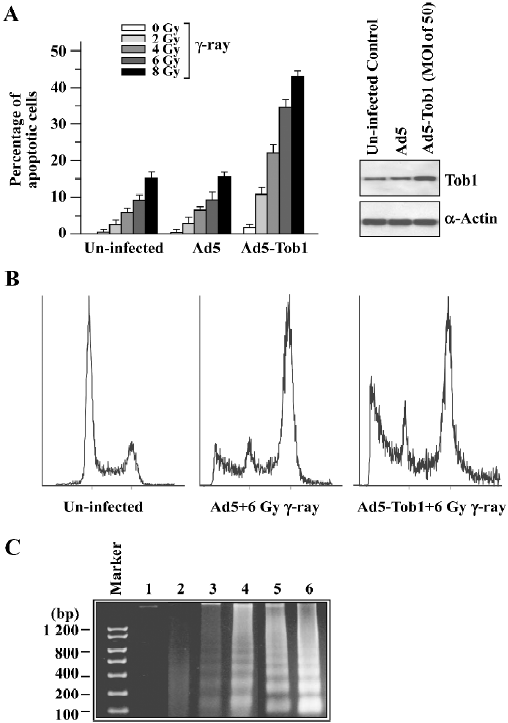
Ad5-Tob1 increases irradiation-induced apoptosis In order to determine whether the increased radiosensitivity by the adenovirus-mediated expression of Tob1 was due to apoptosis, the TUNEL assay using the APO-BRDUTM kit (Phoenixflow, San Diego CA, USA) was performed. The γ-ray irradiation or Ad5-Tob1 (at the MOI of 100 at 48 h infection) alone was ineffective in inducing apoptosis in MDA-MB-231, a cell line having a mutant p53 gene (Figure 3A). However, γ-ray potently induced apoptosis in the presence of 48 h-pre-infection of Ad5-Tob1. This degree of sensitization appeared to be greater than additive. For example, 6 and 8 Gy irradiation alone caused about 9% and 15% apoptosis induction, respectively, and Ad5-Tob1 alone induced approximately 4% apoptosis. The combination, however, induced approximately 35% and 43% of apoptotic cells (radiation alone vs the combination of Ad5-Tob1 and radiation, P<0.01). This synergistic effect was not observed after the combination of Ad5 and γ-ray. A significantly increased induction of apoptosis was also seen by using the DNA fragmentation assay in cells treated with Ad5-Tob1 (at the MOI of 100 at 48 h pre-infection) and γ-ray irradiation (Figure 3B). These results clearly indicated that the increased expression of Tob1 may enhance the apoptosis induction in cells exposed to γ-ray.
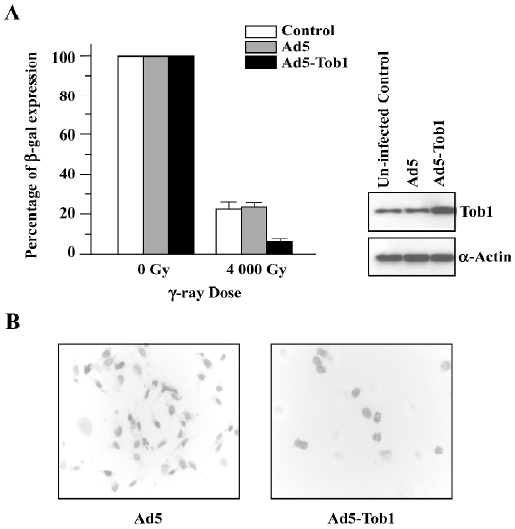
Ad5-Tob1 represses DNA repair Based on the large reduction in Dq seen in the survival curves (Figure 2B), a host cell reactivation assay was carried out to determine whether the radiosensitizing effect of Ad5-Tob1 could be explained as a suppression of the facility to repair DNA damages in MDA-MB-231 cells. To do so, the cells were accepted at 48 h-pre-infection with either Ad5-Tob1 or Ad5 at the MOI of 100 and then infected with Ad-β-gal that had been irradiated with γ-ray irradiation at a single dose of 4000 Gy. The ability of the MDA-MB-231 cells to reactivate the irradiated Ad-β-gal based on β-gal expression was assessed 24 h later. The results of these experiments (Figure 4), presented as the percentage of the controls using unirradiated Ad-β-gal, showed that MDA-MB-231 cells infected with Ad5-Tob1 had a significantly lower ability to reactivate irradiated Ad-β-gal compared with MDA-MB-231 cells that had either uninfection or Ad5 infection (P<0.01). These results indicated that an increase of Tob1 expression by the adenovirus-mediated Tob1 gene transfer results in a reduced capability of MDA-MB-231 to repair DNA.
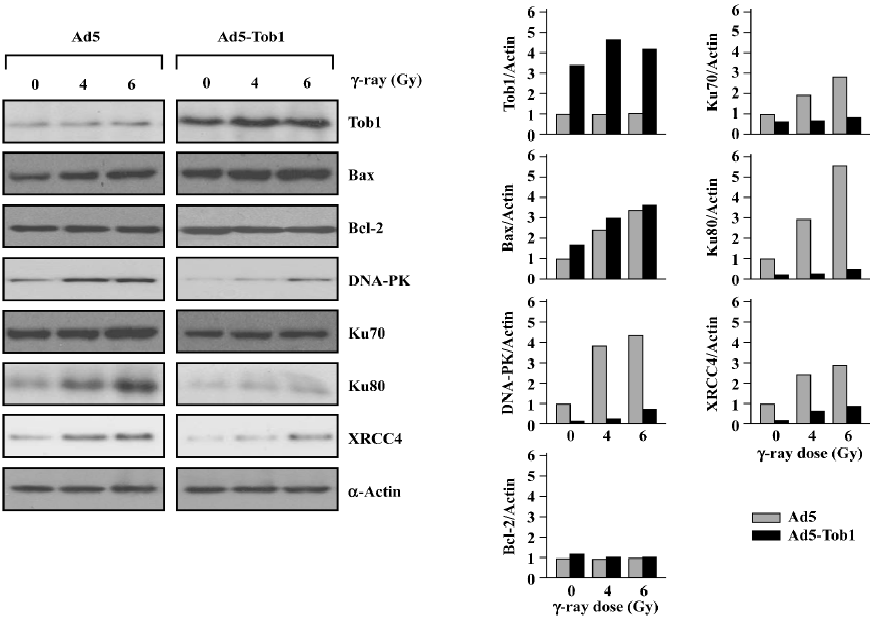
Ad5-Tob1 increases Bax and decreases DNA repair protein expression First, we determined the effect of Ad5-Tob1 on the expression of the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2, 2 well-known members of the Bcl-2 gene family in apoptosis induction[24]. As shown in Figure 5, a significant elevation of the Tob1 protein was observed in the MDA-MB-231 cells infected with Ad5-Tob1. Furthermore, a slight increase of Tob1 was found in response to irradiation with γ-ray, although the significance of such a response is unclear so far. Second, exposure to γ-ray caused a dose-dependent increase of the Bax protein in the cells infected with control Ad-5. The infection of Ad5-Tob1 alone increased the base level of the endogenous Bax protein. Moreover, it enhanced an increase of the γ-ray-mediated Bax, which also showed a γ-ray dose-dependent promotion. However, neither γ-ray+Ad5 nor Ad5-Tob1+γ-ray produced any significant effects on the Bcl-2 protein. Third, we found that Ad5-Tob1 not only reduced the endogenous levels of several proteins involved in the DNA DSB repair, including DNA-PK (Ku70 and Ku80) and XRCC4, but it also blocked the increase of these proteins by γ-ray irradiation (Figure 5). Taken together with the in vitro DNA repair data in Figure 4, these results indicated that the inhibition of DNA repair by Ad5-Tob1 may be due to Ad5-Tob1 downregulation of DNA DSB repair proteins. Further investigation is underway to determine the vital roles of the DNA DSB repair proteins in Tob1-mediated radiosensitivity in our laboratory.
Discussion
Tob1 is an antiproliferative protein and plays a role in preventing cell cycle progression by downregulating the expression and promoter activity of cyclin D1, an important G1 checkpoint player[8,9]. Furthermore, knocking-down or loss of the Tob1 gene in mice results in multiple tumor development[10] and tumor progression[11]. Based on these studies, strategies have been developed for manipulating the Tob1 expression in the treatment of human cancer; one such approach involves the replacement of Tob1 into tumor cells by gene therapy[13].
The present study examined whether Tob1 might radiosensitize breast cancer cells and also the underlying mechanism(s) by examining the repair of radiation-induced DNA damage. Ad5-Tob1 significantly enhanced the radiosensitivity of human breast cancer MDA-MB-231 cells in a synergistic manner as determined based on clonogenic survival compared with the control Ad5 vector (Figure 1). Ad5-Tob1 also restored radiation-induced apoptosis in MDA-MB-231 cells as shown in Figure 2. Therefore, enhanced radiosensitivity may be due to the restoration of radiation-induced apoptosis in some settings.
Consistent with the enhanced induction of apoptosis by a combination of Ad5-Tob1 and γ-ray, we also found that the expression of the pro-apoptotic protein Bax, one of the Bcl-2 family proteins, was increased in MDA-MB-231 cells treated with Ad5-Tob1+γ-ray (Figure 5). It is well known that members of the Bcl-2 family are the most prominent mediators of apoptosis induction by a large number of antitumor drugs and ionizing radiation in a variety of cell types, including cancer cells[25–28]. The Bcl-2 family consists of a growing number of proteins, containing 4 conserved Bcl-2 homology domains (BH, BH2, BH3, and BH4) with a transmembrane domain and can be classified as 3 subgroups[27]: (i) the Bcl-2 subgroup, including all anti-apoptotic proteins, like Bcl-2 and Bcl-xL; (ii) the Bax subgroup, consisting of pro-apoptosis members, like Bax and Bad; and (iii) the third subgroup, containing BH3-only proteins, like Bid and Bim. The last subgroup members interact with either anti-apoptotic proteins or pro-apoptosis members and cause apoptosis promotion. Thus, the upregulation of the pro-apoptosis protein Bax by Ad5-Tob1 may be an important mechanism contributing to the Adt-Tob1 promotion of radiation-mediated apoptosis, although the anti-apoptotic protein Bcl-2 is unaltered. Experiments to determine the possible involvement of other Bcl-2 family members in Ad5-Tob1 radiosensitivity are underway in our laboratory, which will provide more information about the role of Bcl-2 family members in Ad5-Tob1-mediated apoptosis in radiosensitivity.
DNA is a major target in damage caused by ionizing radiation. DNA DSB are potent inducers of mutations and of cell death by irradiation. For the survival of irradiated mammalian cells, the repair of radiation-induced DSB is essential and such repair apparently involves 2 mechanistically distinct, but always overlapping, pathways: HR and NHEJ. The repair of radiation-induced DSB can be detected by using pulsed-field gel electrophoresis techniques, but this approach is not sensitive enough for some types of misrepaired lesions. Thus, we performed the assays of DNA repair by using a host cell reactivation assay. This relatively old approach[29] was updated to incorporate the expression of a reporter gene as the readout; radiation-induced DNA lesions in the reporter gene are effectively and efficiently repaired by the host cells with complete fidelity in order for functional gene expression to be restored. The results shown in Figure 4 indicate that the cells infected with Ad5-Tob1 have significantly less capability to restore reporter gene expression using an irradiated Ad-β-gal vector by more than 60% when compared with controls (uninfected and Ad5 infected). Thus, the overexpression of Tob1 in this context sensitizes cells to ionizing radiation by suppressing their capacity for repairing radiation-induced DNA lesions. Consistent with these DNA repair results, we demonstrated that the expression of certain critical proteins involved in DNA damage repair pathways were downregulated by Ad5-Tob1 in MDA-MB-231 cells in response to ionizing radiation, as shown in Figure 5, that is, both the endogenous base level of DNA-PK (Ku70 and Ku80) and the XRCC4 proteins, and the radiation-increased expressions of these proteins were downregulated and blocked by adenovirus-mediated Tob1 gene expression. It is known that the DNA–PK, activated by DNA ends, is a central component in the NHEJ pathway. DNA-PK is a serine/threonine kinase that consists of a large catalytic subunit of (DNA-dependent protein kinase catalytic subunit, DNA-PKcs) and a DNA-targeting component Ku (a heterodimer of Ku70 and Ku80)[30,31]. Mammalian cells deficient in DNA-PKcs (Ku70 and Ku80) and XRCC4 exhibit hypersensitivity to radiation, deficiencies in DNA DSB repair and V(D)J name is too long to writerecombination[32–34]. Thus, our present studies suggest that Tob1 may participate in DNA DSB repair intermediates and pathways, resulting in cellular hyposensitivity to radiation, although the interplay of these molecular events is not clearly understood at present.
In summary, we have for first time demonstrated that the enhanced expression of Tob1 by the adenovirus-mediated Tob1 gene transfer augments the response of breast cancer cells to ionizing radiation by increasing apoptosis, reducing DNA repair ability, increasing the pro-apoptotic protein Bax, and suppressing the expression of critical proteins in the DNA repair pathway. A complete understanding of these effects must await future studies. However, our present in vitro observations underscore the need for the continued development of strategies for sensitizing human tumor cells to cancer radiotherapy that kill cells by using the adenovirus-mediated expression of Tob1 in clinics.
References
- Kufe D, Weichselbaum R. Radiation therapy: activation for gene transcription and the development of genetic radiotherapy-therapeutic strategies in oncology. Cancer Biol Ther 2003;2:326-9.
- Robson T, Worthington J, McKeown SR, Hirst DG. Radiogenic therapy: novel approaches for enhancing tumor radiosensitivity. Technol Cancer Res Treat 2005;4:343-61.
- Freyta SO, Kim JH, Brown SL, Barton K, Lu M, Chung M. Gene therapy strategies to enhance the effectiveness of cancer radiotherapy. Curr Opin Mol Ther 2004;6:513-24.
- Lumniczky K, Safrany G. Cancer gene therapy: combination with radiation therapy and the role of bystander cell killing in the anti-tumor effect. Pathol Oncol Res 2006;12:118-24.
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002;23:687-96.
- Han Z, Wang H, Hallahan DE. Radiation-guided gene therapy of cancer. Technol Cancer Res Treat 2006;5:437-44.
- Ferguson DO, Alt FW. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene 2001;20:5572-9.
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell 2002;109 Suppl:S45-55.
- Matsuda S, Kawamura-Tsuzuku J, Ohsugi M, Yoshida M, Emi M, Nakamura Y, et al. Tob, a novel protein that interacts with p185erbB2, is associated with anti-proliferative activity. Oncogene 1996;12:705-13.
- Suzuki T. K-Tsuzuku J, Ajima R, Nakamura T, Yoshida Y, Yamamoto T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev 2002;16:1356-70.
- Yoshida Y, Nakamura T, Komada M, Satoh H, Suzuki T, Tsuzuku JK, et al. Mice lacking a transcriptional corepressor Tob are predisposed to cancer. Genes Dev 2003;17:1201-6.
- Ito Y, Suzuki T, Yoshida H, Tomoda C, Uruno T, Takamura Y, et al. Phosphorylation and inactivation of Tob contributes to the progression of papillary carcinoma of the thyroid. Cancer Lett 2005;220:237-42.
- Yanagie H, Sumimoto H, Nonaka Y, Matsuda S, Hirose I, Hanada S, et al. Inhibition of human pancreatic cancer growth by the adenovirus-mediated introduction of a novel growth suppressing gene, tob, in vitro. Adv Exp Med Biol 1998;451:91-6.
- Sasajima H, Nakagawa K, Yokosawa H. Antiproliferative proteins of the BTG/Tob family are degradated by the ubiquitin-proteasome system. Eur J Biochem 2002;269:3596-604.
- Hiramatsu Y, Kitagawa K, Suzuki T, Uchida C, Hattori T, Kikuchi H, et al. Degradation of Tob1 mediated by SCFSkp2-dependent ubiquitination. Cancer Res 2006;66:8477-83.
- Xia F, Powell SN. The molecular basis of radiosensitivity and chemosensitivity in the treatment of breast cancer. Semin Radiat Oncol 2002;12:296-304.
- Suzuki T, Tsuzuku JK, Ajima R, Nakamura T, Yoshida Y, Yamamoto T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev 2002;16:1356-70.
- Cho HN, Lee YJ, Cho CK, Lee SJ, Lee YS. Downregulation of ERK2 is essential for the inhibition of radiation-induced cell death in HSP25 overexpressed L929 cells. Cell Death Differ 2002;9:448-56.
- Kawate H, Wu Y, Ohnaka K, Nawata H, Takayanagi R. Tob proteins suppress steroid hormone receptor-mediated transcriptional activation. Mol Cell Endocrinol 2005;230:77-86.
- Toillon RA, Magne N, Laios I, Lacroix M, Duvillier H, Lagneaux L, et al. Interaction between estrogen receptor alpha, ionizing radiation and (anti-) estrogens in breast cancer cells. Breast Cancer Res Treat 2005;93:207-15.
- Paulsen GH, Strickert T, Marthinsen AB, Lundgren S. Changes in radiation sensitivity and steroid receptor content induced by hormonal agents and ionizing radiation in breast cancer cells in vitro. Acta Oncol 1996;35:1011-9.
- Fan S, Wang J, Ma Y, Yuan R, Meng Q, Erdos M, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001;22:77-87.
- Fan S, Wang J, Yuan R, Rockwell S, Andres J, Zlatapolskiy A, et al. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA damaging-agents. Oncogene 1998;17:131-41.
- Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005;103:1551-60.
- Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 2006;13:1339-50.
- Adams JM, Cory S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem Sci 2001;26:61-6.
- Thomadaki H, Scorilas A. BCL-2 family of apoptosis-related genes: functions and clinical implications in cancer. Crit Rev Clin Lab Sci 2006;43:1-67.
- Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005;103:1551-60.
- Rainbow AJ. Repair of radiation-induced DNA breaks in human adenovirus. Radiat Res 1974;60:155-64.
- Featherstone C, Jackson SP. Ku, a DNA repair protein with multiple cellular functions? Mutat Res 1999;434:3-15.
- Tuteja R, Tuteja N. Ku autoantigen: a multifunctional DNA-binding protein. Crit Rev Biochem Mol Biol 2000;35:1-33.
- Kurimasa A, Ouyang H, Dong LJ, Wang S, Li X, Cordon-Cardo C, et al. Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc Natl Acad Sci USA 1999;96:1403-8.
- Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, et al. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature (Lond) 1996;382:551-5.
- Ouyang H, Nussenzweig A, Kurimasa A, Soares VC, Li X, Cordon-Cardo C, et al. Ku70 is required for DNA repair but not for TCR gene recombination in vivo. J Exp Med 1997;186:921-9.