
Effects of scutellarin on PKCγ in PC12 cell injury induced by oxygen and glucose deprivation
Introduction
Neuronal injury, which is induced by cerebral ischemia/reperfusion, is a very complex process associated with excitotoxicity, oxidative stress, apoptosis, and variations in gene expression or the activation of kinase[1]. Scutellarin (Scu), a known flavone glycoside, is a major active ingredient in breviscapine, which is a mixture extracted from the Chinese herb, Erigeron breviscapus. Scu is used in clinics to treat ischemic cerebrovascular diseases in China (Figure 1).
It is reported that Scu has protective effects against neuronal damage induced by chemical hypoxia or hydrogen peroxide through interaction with a wide variety of targets, including antioxidative action and attenuating neuron damage[2,3]. There is no sufficient evidence proving the effects of Scu against ischemia/reperfusion injury, especially at molecular and cellular level. Therefore, we used the oxygen and glucose deprivation followed by reperfusion (OGD-Rep) model, an in vitro model of cerebral ischemia/reperfusion, to investigate the effects of Scu on protein kinase C (PKC)γ and PC12 cell injury.
PKCγ, a serine/threonine kinase expressed exclusively in neurons of the brain and spinal cord, is activated by Ca2+, diacylglycerides (DAG) and free fatty acids (FFA)[4]. Therefore, this isozyme plays a central nervous system-specific role in mediating the response to ischemic/reperfusion injury. Ischemia causes extensive membrane phospholipids hydrolysis with accumulation of FFA, DAG, and other breakdown products[5]. The changes in membrane phospholipids may modulate the function of membrane proteins and also shift the distribution of PKCγ. Furthermore, during ischemia, irreversible processes including mitochondrial collapse, rapid energy depletion, and ion pump failure result in large increases in intracellular calcium, leading to the activation and increased expression of PKCγ[6,7]. Multiple reports now suggest that PKC may be involved in a positive feedback loop to potentiate NMDAR activity, worsening calcium loading, mitochondrial dysfunction, and promoting cell death[8,9]. So as reported earlier, PKCγ is most effectively translocated to membranes and the nucleus during cerebral ischemia, and plays an important role in neuronal damage[10]. Previous studies have suggested that breviscapine inhibits the activity of PKC. So we estimated that the protective effects of Scu on neuron apoptosis might be related to this inhibitive effect on PKC, especially on PKCγ [11,12].
PC12 cells have been extensively used as an in vitro model system to study the mechanisms of neuronal cell death after ischemic insult, and to develop potential neuroprotective agents. Ischemic stroke causes a significant amount of cell damage resulted from an insufficient supply of glucose and oxygen to brain tissues[13,14]. PC12 cells are subjected to an initial short phase of OGD-Rep. The initial phase of OGD mimics the lack of oxygen and glucose supply, while the prolonged phase of OGD-Rep reflects the reperfusion of oxygen and glucose supply to the injured brain[15,16].
In the present study, we investigated whether Scu, the major ingredient of Erigeron breviscapus, had neuronal protective effects against ischemia/reperfusion injury by influencing PKCγ. So we used undifferentiated rat pheochromocytoma PC12 cells and the OGD-Rep model to study the effects of Scu on cell survival, apoptosis, [Ca2+]i, and the expression and activation of PKCγ.
Materials and methods
Chemicals and drugs Scu (purity >92%) was supplied by Kunming Institute of Botany, Chinese Academy of Science (Kunming, Yunnan, China). Fura-2 acetoxymethyl ester (Fura-2AM) and propidium iodide (PI) staining solution were purchased from BD PharMingen (San Diego, CA, USA). TRIzol Reagent was purchased from Invitrogen (Carlsbad, CA, USA). M-MLV reverse transcriptase was purchased from Promega (Madison, WI, USA). Diphenyl-tetrazolium bromide (MTT) and monoclonal anti-protein kinase Cγ (PKCγ) was purchased from Sigma (St Louis, MO, USA). The SuperSignal West Pico Trial Kit was purchased from Pierce (Rockford, MA, USA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Carlsbad, CA USA). LDH activity kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals and solvents in experiments were of analytical grade.
Cell culture The PC12 cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA,USA) and maintained at 37 °C in a humidified atmosphere containing 5% CO2 in high glucose DMEM supplemented with 10% heat-inactivated fetal calf serum, 5% heat-inactivated horse serum, 100 kU/L penicillin, and 100 mg/L streptomycin.
PC12 ischemic model The PC12 cells were grown as monolayers in tissue culture flasks under normal conditions. To initiate OGD, the cell culture medium was removed and the cells were washed twice with glucose-free DMEM, and incubated in the same volume glucose-free DMEM containing Scu (10, 30, and 100 μmol/L). The cell culture was incubated at 37 °C in an oxygen-free incubator (95% N2 and 5% CO2) for 4 h (OGD). Then the OGD glucose was added to attain a final concentration of 4.5 mg/L, followed by incubation for 20 h (OGD-Rep) in normal conditions[17]. Scu was freshly prepared as stock solution with dimethyl sulfoxide (DMSO) and diluted with glucose-free DMEM. Less than 0.1% DMSO has no protective or toxic effects when used alone.
Assay of cell survival and cell damage Cell survival was evaluated by MTT assay. After reperfusion, MTT solution in phosphate-buffered saline was added to attain a final concentration of 0.5 g/L, then incubation continued for 4 h. Finally, the media was removed and an equal volume of dimethylfuramide was added. After the mixtures were kept for 10 min, the amount of MTT was quantified by determining its absorbance at 570 nm using a Spectra MAX 190 micro-plate reader (Molecular Devices Inc, USA). The amount of LDH released by the cells was determined using an LDH activity assay kit according to manufacturer’s instructions. LDH activity is proportional to the rate of pyruvate loss, which was measured at 440 nm. LDH activity in the cells was determined after incubation in a hypotonic solution containing 1% Triton X-100, at pH 7.4.
Flow cytometric detection of apoptosis cells The cells were collected and resuspended in ice-cold ethanol (70%), then fixed at 4 °C for 24 h and resuspended in 1 mL DNA staining reagent (RNase 50 mg/mL, 0.1% Triton X-100, 0.1 mol/L EDTA, and 50 mg/mL PI). Red fluorescence was detected by using a FACS 440 flow cytometer (Becton Inc, USA). Ten thousand cells in each sample were analyzed and the percentage of apoptotic cell accumulation in the sub-G1 peak was calculated.
Evaluation of DNA laddering The total DNA was extracted according to a previous study[18]. The DNA pellet was resuspended in loading buffer (10 mmol/L Tris, 1 mmol/L EDTA, 5% glycerol, and 0.25% bromophenyl blue, pH 7.5), and the entire sample was then subjected to electrophoresis on a 2% agarose gel using a TBE running buffer (89 mmol/L Tris, 89 mmol/L boric acid, and 2 mmol/L EDTA, pH 8.3) containing 0.5 g/L ethidium bromide. The fluorescence intensity of the DNA bands was measured using the Bio-Rad Molecular Imaging System (Bio-Rad, USA).
Measurement of [Ca2+]i After reperfusion, the PC12 cells were loaded with 5 mmol/L (final concentration) Fura-2AM, 0.1% DMSO and 1% BSA for 30 min at room temperature in dark conditions, then in a humidified incubator for 30 min at 37 °C. Fluorescence measurement was carried out with an F-4500 fluorescence spectrophotometer (Hitachi, Japan). Fura-2AM-loaded PC12 cells were exposed sequentially to an excitation wavelength of 340 nm and 380 nm (bandwidth 10 nm), and the emission signal was monitored at a wavelength of 510 nm (bandwidth 10 nm). Fluorescence ratios were converted into calcium concentrations by using the following equation: [Ca2+]i=Kd (R-Rmin)/(Rmax-R)B.
Preparation of total RNA and semiquantitative RT-PCR The cells were collected and the total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions. RNA concentration was estimated by absorbance at 260 nm using a UV spectrophotometer. The total RNA of each sample was reverse-transcribed into cDNA using the reverse transcription system. The cDNA was amplified with the following specific primers: PKCγ: 5'-TTG ATG GGG AAG ATG GGG AGG-3' (upstream) and 5'-GAA ATC AGC TTG GTC GAT GCT G-3' (downstream). Amplifications were performed as follows: 30 cycles, at 94 °C for 30 s, 58 °C for 1 min, and 72 °C for 1 min. The PCR products were normalized in relation to standards of GAPDH mRNA.
Preparation of total PKCγ protein and subcellular fractionation The cells were collected, and the total PKCγ protein was extracted using TRIzol reagent according to the manufacturer’s instructions. The subcellular fraction of PKCγ was extracted according to a previous study[19]. Twenty µL of each sample was solubilized with NaOH (1 mol/L) for protein concentration determination.
Polyacrylamide gel electrophoresis and Western blot analysis Twenty µg protein per lane was subjected to 10% SDS-PAGE electrophoresis for 2 h. Resolved proteins were electroblotted onto NC membranes for 2 h at 250 mA. The blots were incubated with TBST (0.1 mol/L Tris buffer, pH 7.4, 0.9% NaCl, and 0.1% Tween 20) containing 5% dry skim milk, followed by incubation with TBS (0.1 mol/L Tris buffer, pH 7.4, 0.9% NaCl) containing anti-PKCγ monoclonal antibodies and 3% BSA for 2 h, and incubation with secondary antibody streptavidin-horseradish peroxidase-conjugated affinity mouse anti-rat IgG (1:10000 dilution) for 2 h. The bound immunoproteins were detected by enhancer chemiluminescent assay, and band intensity was quantified with a densitometric scanner.
Statistical analysis Data were expressed as mean±SD. Statistical analysis was performed by ANOVA, with P<0.05 as the significant level.
Results
Effect of Scu on PC12 cell viability and cell damage The effect of Scu alone on cell viability was studied using MTT assay. The results showed Scu (5–200 µmol/L) had no effects on PC12 cells. After OGD-Rep, PC12 cell viability decreased significantly (<20%) and LDH release increased from 6.77% to 12.30%. The addition of Scu at the concentration of 10–100 µmol/L significantly attenuated the decrease of cell viability and the increase of LDH release. LDH leakage was expressed as the percentage of the total LDH activity (LDH in the medium+LDH in the cells), according to the following equation: %LDH=(LDH activity in the medium/total LDH activity)×100 (Table 1).
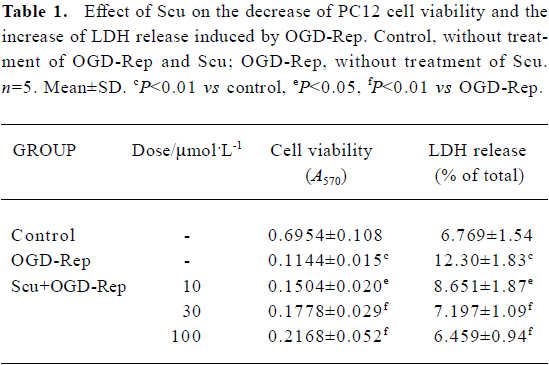
Full table
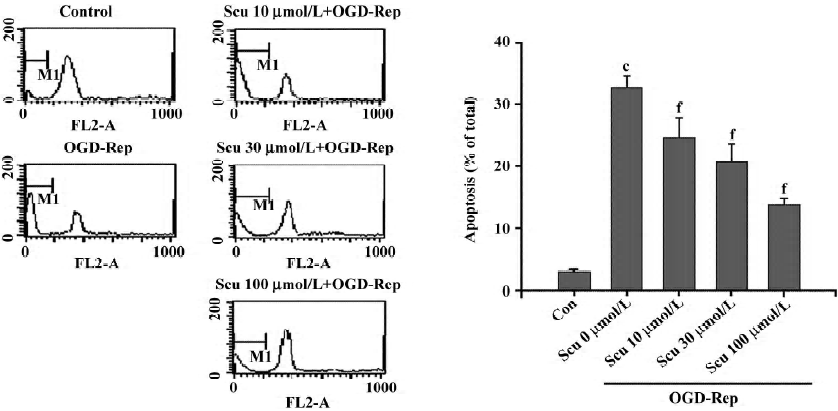
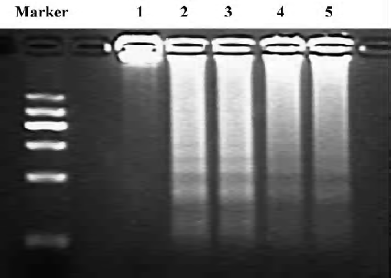
Effect of Scu on cell apoptosis The sub-G1 peak in the flow cytometry detection was considered an indicator of cell apoptosis. The data was presented as the percentage of apoptotic cells. OGD-Rep significantly induced the sub-G1 peak, indicating an apoptotic cell accumulation of 33.34%. The addition of Scu at the concentration of 10–100 µmol/L significantly attenuated OGD-Rep-induced apoptosis (25.88%, 21.67%, and 13.85%; Figure 2). The data is in agreement with the results of the DNA ladder analysis (Figure 3).
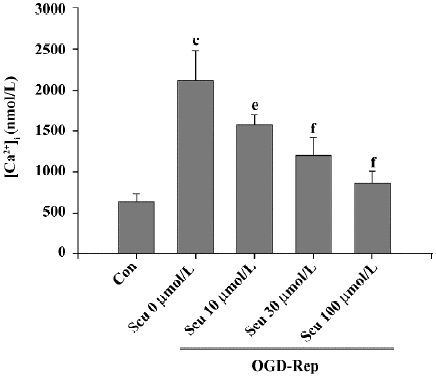
Effect of Scu on [Ca2+]i OGD-Rep induced a significant increase in the resting levels of [Ca2+]i from 646±92 to 2112±376 nmol/L. This increase was significantly inhibited by Scu at 10, 30, and 100 µmol/L (Figure 4).
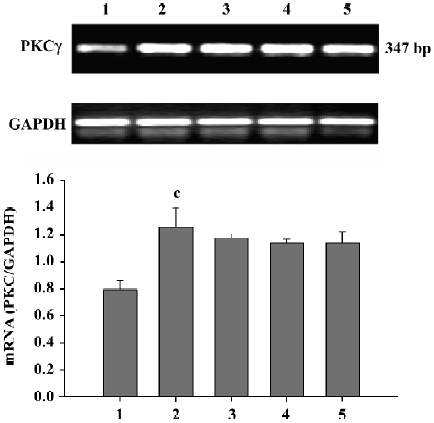
Effect of Scu on PKC mRNA expression in PC12 cells After OGD-Rep, there was a significant increase in PKCγ mRNA expression. At the concentration of 10–100 µmol/L, Scu was less potent in antagonizing the OGD-Rep-induced increase of PKCγ mRNA expression (P>0.05; Figure 5).
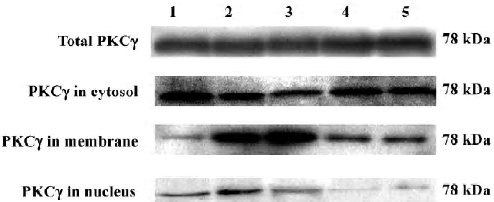
Effects of Scu on protein expression and subcellular distribution of PKCγ in PC12 cells The translocation of PKCγ from cytosol to the membrane and the nucleus is a hallmark of PKCγ activation. Western blotting experiments were performed to detect the protein level of total PKCγ proteins and PKCγ in cytosol, the membrane, and the nucleus of PC12 cells. Our results showed that neither reperfusion nor treatment of Scu changed the expression of PKCγ at protein level (P>0.05), but in OGD-Rep injury, the protein level of PKCγ decreased in cytosol and increased in the membrane and nucleus significantly. Scu at the concentration of 100 μmol/L significantly attenuated the decrease of the protein level of PKC in cytosol and the increase of the protein level of PKCγ in the membrane and nucleus, whereas Scu at the concentration of 30 μmol/L only attenuated the increase of the protein level of PKCγ in the membrane and nucleus (Figure 6; Table 2).
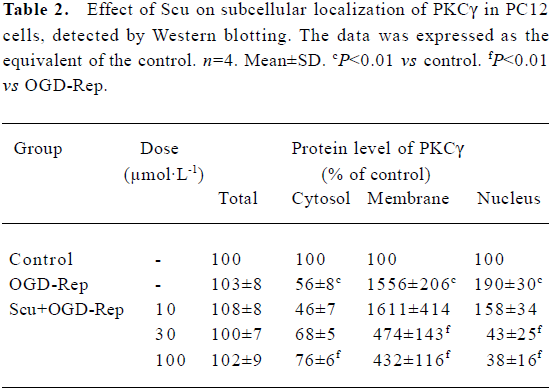
Full table
Discussion
Stroke occurs when local thrombosis, embolic particles, or the rupture of blood vessels interrupts the blood flow to the brain. Excitotoxicity, oxidative stress, and apoptosis propagates through a distinctive and mutually-exclusive signal transduction pathway and contributes to neuronal injury following ischemia/reperfusion[20]. In the present study, we demonstrated that Scu (10–100 µmol/L) significantly increased cell survival (by MTT assay) and reduced LDH release after OGD-Rep. Therefore, Scu has a protective effect against PC12 cell injury induced by OGD-Rep.
Apoptosis that often occurs during the developmental process is an additional route to pathological neuronal death in the mature nervous system during ischemia/reperfusion, and drugs inhibiting those changes attenuate neuronal injury following ischemia/reperfusion[21,22]. The alteration of various apoptosis-associated genes and proteins, calcium imbalance, and oxidative stress contribute to apoptosis during ischemia/reperfusion. The results in our study indicate that the OGD-Rep-induced PC12 cell death, in part due to apoptosis and Scu (10–100 µmol/L), decreased the rate of apoptosis. Therefore, it is tempting to suggest that the neuroprotective effect of Scu is related to a reduction in apoptosis following ischemia/reperfusion.
Ca2+ is a ubiquitous intracellular messenger, the intracellular concentration of which is tightly regulated in order to efficiently control signaling mechanisms[23,24]. The selective accumulation of intracellular Ca2+ following OGD-Rep injury can cause neuronal apoptosis through a mechanism that involves the excessive release of glutamate and the activation of several calcium-dependent/activated enzymes such as proteases, protein kinases, phospholipases, NO syn-theses, and endonucleases[25,26]. Our study shows that OGD-Rep can significantly elevate the level of intracellular Ca2+. This elevation could be attenuated by Scu at concentrations of 10–100 µmol/L. Previous studies in our laboratory on the direct effects of Scu on calcium using the intracellular cation measurement system, with the help of the Ca2+-indicator Fura-2AM, show that Scu had no inhibitory effect on the increase of [Ca2+]i induced by ATP and high K+ in PC12 cells. It was reported that Scu could scavenge radicals and inhibit lipid peroxidation, so we thought the inhibitive effect of Scu on [Ca2+]i might be related to its antioxidation activity[11]. PKCγ is activated by Ca2+, and the activation of PKCγ also increases intracellular Ca2+. Therefore, Scu might influence PKCγ by decreasing intracellular Ca2+ following OGD-Rep.
PKC is activated by stroke and plays a damaging role during stroke. The mechanisms of PKC activation are multifactorial: ischemia/reperfusion induces an influx of calcium and sodium into the cell, leading to the release of intracellular calcium stores, lipid peroxidation, and the generation of free radicals[27,28]. These events lead to increased expression and activation of multiple PKC isozymes. It was reported that the expression of PKCγ increases over a period of postischemic reperfusion[29]. However, other studies showed that the expression of PKCγ decreased during an extended reperfusion[30]. It was estimated that the alteration of the expression depended on the ischemic model and the period of reperfusion used in the studies. A previous study also showed that Scu could inhibit PKCγ activity in the rat MCAO model. PKCγ activity is regulated by 2 distinct mechanisms: by phosphorylation, which regulates the active site, and subcellular localization of the enzyme; and by second messengers (Ca2+ and DAG) which promote PKCγ membrane association. In our previous study, we found that Scu inhibited the increase of [Ca2+]i and oxidative stress, both of which influence the translocation of PKCγ after OGD-Rep, so we guess that the Scu inhibited the activity of PKCγ through inhibiting the translocation of PKCγ by influencing the cofactors Ca2+ and DAG. The results showed that the expression of PKCγ increased significantly following OGD-Rep injury. Scu had no significant effect on this elevation at concentrations of 10–100 µmol/L (P>0.05), but the results of Western blotting showed that the total PKCγ protein did not change after OGD-Rep. It is thought that the membrane-bound PKCγ, compared to the cytosolic form, is more sensitive to proteolysis, thus the degradation of PKCγ increased during reperfusion because of the activation of proteinase by the elevation of [Ca2+]i. Although reperfusion increased the expression of PKCγ at the mRNA level, it did not alter the expression of PKCγ at the protein level following OGD-Rep. Selective changes in the redistribution of PKCγ occurred after ischemic injury and reperfusion, but those changes in PKCγ activity are still not clear. Several reports have demonstrated that PKCγ becomes inactive and down-regulated, whereas others report that PKCγ is activated specifically during reperfusion[19,31]. Activation of the PKCγ leads not only to their translocation from cytosol to the plasma membrane, but also to various intracellular membranes and to the cell nucleus[32,33]. Nuclear translocation of PKCγ is of particular importance because it may enable PKCγ to exert a direct role in nuclear signaling by phosphorylating nuclear substrates, including DNA-modifying enzymes or transcription factors. In our study, there was a significant increase of PKCγ in both the membrane and nucleus, accompanied with a significant decrease in cytosol following OGD-Rep-induced injury. Scu significantly attenuated the translocation of PKCγ to the membrane and nucleus at the concentration of 30 and 100 µmol/L. Scu also significantly attenuated the decrease in cytosol at the concentration of 100 µmol/L. The data showed that Scu, which attenuated the increase of [Ca2+]i and oxidative stress, could protect PC12 cells against OGD-Rep-induced injury as a PKCγ inhibitor through inhibiting the activation of PKCγ, rather than the expression of PKCγ. However, there is no evidence that the effect of Scu on PKCγ is related to phosphorylation, the key step of the translocation of PKCγ. Further investigation is needed to show the relationship between Scu and phosphorylation.
In conclusion, these results demonstrate that due to its antioxidation activity against ischemia/reperfusion-induced oxidative stress, Scu attenuates the increase of [Ca2+]i and then inhibits the translocation of PKCγ, leading to a decrease in the rate of apoptosis, and possessing a significant protective effect against OGD-Rep-induced injury in PC12 cells.
References
- Seok JW, Doo YK, Byoung JG. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol 2002;35:67-86.
- Hong H, Liu GQ. Protective against hydrogen peroxide-induced cytotoxicity in PC12 cells by scutellerin. Life Sci 2004;74:2959-73.
- Hu XM, Zhou MMOL, Zeng FD. Neuroprotective effects of scutellarin on rat neuronal damage induced by cerebral ischemia/ reperfusion. Acta Pharmacol Sin 2005;26:1454-9.
- Saito N, Shirai Y. Protein kinase C gamma (PKC gamma): function of neuron specific isotype. J Biochem (Tokyo) 2002;132:683-7.
- Bazan NG. Free arachidonic acid and other lipids in the nervous system during early ischemia and after electroshock. Adv Exp Med Biol 1976;72:317-35.
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999;79:1431-568.
- Matsumoto S, Shamloo M, Matsumoto E, Isshiki A, Wieloch T. Protein kinase C-gamma and calcium/calmodulin-dependent protein kinase II-alpha are persistently translocated to cell membranes of the rat brain during and after middle cerebral artery occlusion. J Cereb Blood Flow Metab 2004;24:54-61.
- Chakravarthy BR, Wang J, Tremblay R, Atkinson TG, Wang F, Li H, et al. Comparison of the changes in protein kinase C induced by glutamate in primary cortical neurons and by in vivo cerebral ischaemia. Cell Signal 1998;10:291-5.
- MacDonald JF, Kotecha SA, Lu WY, Jackson MF. Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Curr Drug Targets 2001;2:299-312.
- Cardell M, Bingren H, Wieloch T, Zivin J, Saitoh T, Saitoh X. Protein kinase C is translocated to cell membranes during cerebral ischemia. Neurosci Lett 1990;119:228-32.
- Yu G, Lou Y, Peng GG, Dong WW. Effect and mechanism of protein kinase C inhibitor on cerebral ischemia in rats. Stroke Nervous Disease 2002;9:321-4.
- Shuai J, Dong WW. Experimental research of PKC inhibitor, erigeron breviscapus on the ischemic/reperfusion brain injury. Chin Pharmacol Bull 1998;14:77-9.
- Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: pathophysiology. J Neurosurg 1992;77:169-84.
- Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part II: mechanisms of damage and treatment. J Neurosurg 1992;77:337-54.
- Abu RS, Trembovler V, Shohami E, Lazarovici P. A tissue culture ischemic device to study eicosanoid release by pheochromocytoma PC12 cultures. J Neurosci Methods 1993;50:197-203.
- Tabakman RP, Lazarovici P, Kohen R. Neuroprotective effects of carnosine and homocarnosine on pheochromocytoma PC12 cells exposed to ischemia. J Neurosci Res 2002;68:463-69.
- Tabakman R, Jiang H, Shahar I, Arien-Zakay H, Levine RA, Lazarovici P. Neuroprotection by NGF in the PC12 in vitro OGD model: involvement of mitogen-activated protein kinases and gene expression. Ann N Y Acad Sci 2005;1053:84-96.
- Oberdoerster J, Kamer AR, Rabin RA. Differential effect of ethanol on PC12 cell death. J Pharmacol Exp Ther 1998;287:359-65.
- Huang HM, Weng CH, Ou SC, Hwang T. Selective subcellular redistributions of protein kinase C isoforms by chemical hypoxia. J Neurosci Res 1999;56:668-78.
- Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg 2005;18:335-50.
- Fukunaga K, Ishigami T, Kawano T. Transcriptional regulation of neuronal genes and its effect on neural functions: expression and function of forkhead transcription factors in neurons. J Pharmacol Sci 2005;98:205-11.
- Fujimura M, Tominaga T, Chan PH. Neuroprotective effect of an antioxidant in ischemic brain injury: involvement of neuronal apoptosis. Neurocrit Care 2005;2:59-66.
- Gill DL, Ghosh TK, Mullaney JM. Calcium signaling mechanisms in endoplasmic reticulum activated by inositol 1,4,5-trisphosphate and GTP. Cell Calcium 1989;10:363-74.
- Carafoli E. Calcium pump of the plasma membrane. Physiol Rev 1991; 71: 129£53.
- Gwag BJ, Canzoniero LM, Sensi SL, DeMaro JA, Koh JY, Goldberg MP, et al. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neuroscience 1999;90:1339-48.
- Verkhratsky A, Toescu EC. Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J Cell Mol Med 2003;7:351-61.
- Katsura K, Kurihara J, Siesjo BK, Wieloch T. Acidosis enhances translocation of protein kinase C but not Ca2+/calmodulin-dependent protein kinase II to cell membranes during complete cerebral ischemia. Brain Res 1999;849:119-27.
- Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem 1995; 270: 28 495–8.
- Savithiry S, Kumar K. The mRNA levels of Ca-independent forms of protein kinase C in postischemic gerbil brain by Northern blot analysis. Mol Chem Neuropathol 1994;21:1-11.
- Zablocka B, Maternicka K, Zalewska T, Domanska-Janik K. Expression of Ca2+-dependent (classical) PKC mRNA isoforms after transient cerebral ischemia in gerbil hippocampus. Brain Res 1998;779:254-8.
- Selvatici R, Marino S, Piubello C, Rodi D, Beani L, Gandini E, et al. Protein kinase C activity, translocation, and selective isoform subcellular redistribution in the rat cerebral cortex after in vitro ischemia. J Neurosci Res 2003;71:64-71.
- Asaoka Y, Nakamura S, Yoshida K, Nishizuka Y. Protein kinase C, calcium and phospholipid degradation Trends Biochem Sci 1992;17:414-7.
- Buchner K. Protein kinase C in the transduction of signals toward and within the cell nucleus Eur J Biochem 1995;228:211-21.