
Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against ischemia-reperfusion injury in rat hearts1
Introduction
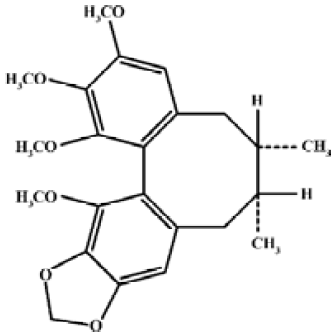
Schisandrin B (Sch B, Figure 1) is the most abundant, active dibenzocyclooctadiene derivative isolated from the fruit of Schisandra chinensis (Turcz) Baill, a traditional Chinese herb clinically used for the treatment of viral and chemical hepatitis[1]. Previous studies in our laboratory have demonstrated the ability of Sch B to protect against myocardial ischemia-reperfusion (I–R) injury[2]. The cardioprotection afforded by Sch B pretreatment were associated with the enhancement in tissue glutathione antioxidant status, particularly in the mitochondrion[2–4]. Recent studies also showed that Sch B protected against myocardial I–R injury partly by inducing heat shock protein (Hsp)25 and Hsp70 expression in rats[5]. However, it is still unclear whether Sch B treatment produces any effect on the sensitivity of the mitochondria to permeability transition (PT). Growing evidence has accumulated suggesting the involvement of PT of the mitochondrial inner membrane in the pathogenesis of oxidant injury in various tissues, including myocardial I–R injury[6,7]. One critical event of mitochondrial PT is the permeability of the inner membrane to small ions and solutes with Mr <1500 daltons, with the consequent, large amplitude swelling of the mitochondria[8]. The opening of mitochondrial PT pores plays an important role in regulating necrotic and apoptotic cell death[9]. While the loss of ion homeostasis resulting from ATP depletion following PT can lead to necrosis[10]. PT also causes the leakage of cytochrome c from the mitochondria to the cytosol[11]. The released cytochrome c can trigger a cascade of events that eventually lead to apoptosis[11,12].
Increased Ca2+, reactive oxidant species (ROS), and inorganic phosphate, as well as decreased membrane potential, have been shown to activate mitochondrial PT, whereas Ca2+ chelator, antioxidants, such as glutathione and ubiquinone analogs, ATP/ADP and other translocase ligands, such as cyclosporin A (CsA), inhibit mitochondrial PT[13,14]. In order to further elucidate the molecular mechanism underlying the cardioprotective action of Sch B, we endeavoured to investigate the effect of Sch B treatment on the sensitivity of the mitochondria to Ca2+-stimulated PT in rat hearts under normal and I–R conditions. Changes in mitochondrial Ca2+ content, ROS production, and cytochrome c release were also examined in relation to alterations in the sensitivity to mitochondrial PT.
Materials and methods
Chemicals and herbal material CsA was purchased from Sigma Chemical Co (St Louis, MO, USA). All other chemicals were of analytical grade. The dried fruits of Schisandra chinensis were imported from mainland China. They was authenticated and supplied by a commercial dealer (Lee Hoong Kee Limited, Hongkong, China) in Hong Kong. Sch B was purified from the petroleum ether extract of Schisandra fruits, with the purity being higher than 95% as determined by HPLC analysis[15].
Animal care Sprague-Dawley rats (8–10 weeks old, weighing 250–300 g) were maintained under a 12 h dark/light cycle at about 22 oC and allowed food and water ad libitum in the Animal Care Facilities at the Hong Kong University of Science and Technology (HKUST, Hong Kong, China). All experimental protocols were approved by the University Committee on Research Practice at HKUST.
Drug treatment The animals were randomly divided into groups of 5 animals. In the pretreatment groups, the rats were treated intragastrically with Sch B (dissolved/suspended in olive oil) at a single dose of 1.0 or 2.0 mmol/kg. Untreated controls received the vehicle only. The hearts were isolated from the rats and subjected to I–R experiment 48 h following the administration of Sch B. Previous studies indicated that the optimal single dose of Sch B for protecting against myocardial I–R injury ranged from 1 to 2 mmol/kg, and the maximum protective effect was observed at 48 h post-dosing with Sch B[3].
Isolated-perfused rat hearts The hearts were excised quickly from the phenobarbital-anesthetized rats and immediately immersed in ice-cold and heparinized (50 unit/mL) saline. The aorta was cannulated and then transferred to a warm, moist chamber of the perfusion apparatus. The hearts was retrogradely perfused, with the coronary flow rate ranging from 8 to 12 min, according to the Langendorff method as described[16].
Myocardial I–R After an initial 30 min of perfusion for equilibration, the isolated hearts were subjected to a 40 min period of “no-flow” global ischemia followed by 20 min reperfusion. Coronary effluent was collected in 1 min fractions at increasing time intervals during the course of equilibration and reperfusion. The fractions were immediately placed on ice until assayed for lactate dehydrogenase (LDH) activity. The extent of LDH leakage during the reperfusion period, an indirect index of myocardial injury, was estimated by computing the area under the curve (AUC) of the graph plotting the percentage of LDH activity (with respect to the mean pre-ischemic value measured during the equilibration period) against the reperfusion time (1–20 min) as describ-ed[4], and the value was expressed as arbitrary units (AU). Non-I–R hearts were perfused for 90 min. After the non-I–R or I–R procedure, ventricular tissue samples were obtained and subjected to biochemical analysis.
Preparation of mitochondrial fractions and cytosolic fractions Myocardial ventricular tissue samples were rinsed with ice-cold isotonic buffer [210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L HEPES, 1 mmol/L EGTA (pH 7.4) 0.2 mg/mL soybean trypsin inhibitor, 0.2 mg/mL bacitracin, and 0.16 mg/mL benzamidine]. Myocardial tissue homogenates were prepared by homogenizing 0.8 g minced tissues in 8 mL ice-cold isotonic buffer. The mitochondrial fractions were prepared by differential centrifugation as described[17]. The homogenates were centrifuged at 600×g for 20 min. After collecting the supernatants, the pellets were resuspended with the same volume of ice-cold homogenizing buffer (without various protease inhibitors) and recentrifuged at 600×g again. The procedure was repeated twice. The pooled supernatants (a total of 4 volumes) were centrifuged at 8000×g for 30 min, and the mitochondrial pellets were collected. The supernatant was saved for the preparation of the cytosolic fraction. The mitochondrial pellets were then washed with the same volume of ice-cold sucrose buffer containing 210 mmol/L mannitol, 70 mmol/L sucrose, and 5 mmol/L HEPES–KOH (pH 7.4) and the mixtures were centrifuged at 8000×g for 30 min. The washing procedure was repeated. The mitochondrial pellets were resuspended in 0.5–1.0 mL ice-cold sucrose buffer and constituted the mitochondrial fractions. The mitochondrial fractions, prepared from various rat tissues by a similar protocol in our laboratory, were found to carry out oxidative phosphorylation in a normal manner[18]. The cytosolic fraction was prepared by centrifuging the above supernatant at 100 000×g for 60 min at 4 oC.
Mitochondrial ROS generation The extent of mitochondrial ROS generation in vitro was estimated as an indirect measure of mitochondrial antioxidant capacity and structural integrity. An aliquot (50 µL) of mitochondrial fraction (50 µg protein/mL) and 60 µL 2',7'-dichlorofluorescin diacetate (DCFDA; Fluka, Switzerland) solution (5 µmol/L in incubation buffer containing 0.1% DMSO) were added into the wells of a black microtiter plate. The mixture was incubated at 37 oC for 10 min under dark condition in a Victor2 Multi-Label Counter (Perkin–Elmer, Wellesley, MA, USA). After the incubation, 50 µL incubation buffer [0.1 mmol/L EGTA, 5 mmol/L KH2PO4, 3 mmol/L MgCl2, 145 mmol/L KCl, 30 mmol/L HEPES (pH 7.4)] and 50 µL substrate solution (20 mmol/L pyruvate and 10 mmol/L malate) were added. Fluorescence intensity (excitation at 485 nm and emission at 535 nm) of the reaction mixture was monitored every 5 min for 30 min. The mitochondrial ROS generation was reflected by the fluorescence intensity of the sample after subtracting the value of a blank sample containing incubation buffer, substrate solution, and DCFDA. The extent of ROS generation over the 30 min period of incubation was estimated by computing the AUC of the graph plotting fluorescent intensity against time (0–30 min) and expressed as an AU.
Mitochondrial PT The measurement of mitochondrial PT was performed according to a procedure modified from Kowaltowski et al[19]. An aliquot (1.6 mL) of the mitochondrial sample (0.5 mg protein/mL) was prepared by mixing the mitochondrial fraction with incubation buffer containing 125 mmol/L sucrose, 65 mmol/L KCl, 10 mmol/L HEPES (pH 7.2), 5 mmol/L freshly prepared succinate, and 5 µmol/L freshly prepared rotenone. Aliquots (200 µL) of the mitochondrial sample were mixed with 10 µL of CsA (5 μmol/L) in 0.5% (w/w) ethanol final concentration or incubation buffer. The mixtures were incubated at 30 oC for 5 min. An aliquot (10 µL) of calcium chloride (Ca2+) solution (1 µmol/L final concentration) was then added, and the mixtures were incubated at 30 oC for 5 min. Aliquots (180 µL) of the mixtures were added into a 96-well microtiter plate, and the initial absorbance of the mixtures at 520 nm was monitored for 5 min at 30 oC. The swelling reaction was then started by adding 20 µL potassium phosphate (0.5 mmol/L, pH 7.2), and the absorbance at 520 nm of the reaction mixtures was read every 2 min for 30 min at 30 oC using the Victor V2 Multi-Label Counter. The extent of mitochondrial swelling was estimated by computing the AUC of the declining graph plotting the percentage of the initial absorbance (100% as baseline) against time (min) to obtain AUC1. The extent of mitochondrial PT (ΔAUC1) was estimated by subtracting the AUC1 with CsA from the AUC1 without CsA. Ca2+-induced mitochondrial PT was expressed in a ratio of ΔAUC1 induced by both Ca2+ and PO43– to that induced by PO43– only.
Mitochondrial Ca2+ content The Ca2+ content was measured using a Ca2+-sensitive fluorescence probe Fluo-5N AM ester (Molecular Probe, OR, USA) by the Victor2 V Multi-Label Counter, as described in Menze et al[20]. The Ca2+ dissociation constant (Kd) was determined by using a Ca2+ calibration kit at a concentration range of 1–1000 µmol/L, with the Kd value estimated at being ~98 µmol/L, which is in agreement with the data provided by the manufacturer. An aliquot (25 µL) of the mitochondrial fraction (0.5 mg/mL final concentration) was mixed with 25 µL incubation buffer (100 mmol/L KCl and 30 mmol/L MOPS, pH 7.2) in a 96-well black microtiter plate. The mixture was incubated at 25 oC for 15 min and then added with 25 µL digitonin (50 µg/mL) and 25 µL Fluo-5N AM ester (1 µmol/L in 0.005% Pluronic F-127). Presumably, by permeating the membrane, digitonin can facilitate the entry of the fluorescence dye into the mitochon-dria. Experimental data indicated that the fluorescence intensity of Fluo-5N measurable in rat heart mitochondrial preparations was increased by 20-fold in the presence of digitonin. The reaction mixture was incubated at 25 oC for 30 min, and the fluorescence reading was measured at the excitation wavelength of 488 nm and emission at 532 nm. The mitochondrial Ca2+ content was estimated from the standard calibration curve and expressed in μmol/mg protein.
Biochemical analysis The LDH activity of coronary effluent was measured as described[4]. The cytosolic cytochrome c level, as an indirect measure of mitochondrial cytochrome release, was estimated by Western blot analysis using specific antibodies of cytochrome c (clone 7H8.2C12, BD PharMingen, San Diego, CA, USA) following SDS–PAGE analysis of the cytosolic fractions using a separating gel of 15% acrylamide. The extent of mitochondrial contamination in the cytosolic fractions was determined using specific antibodies against complex IV. The immunoblots were visualized by enhanced chemiluminescence reaction (Amersham ECL+, Piscataway, NJ, USA) and analyzed by densitometry (Bio-Rad). The amounts (AU) of cytochrome c were normalized with reference to actin levels (AU) in the sample. The protein concentrations of the mitochondrial fractions were determined using a Bio-Rad protein assay kit (Hercules, CA, USA).
Statistical analysis Data were analyzed by one-way ANOVA. Post-hoc tests for pair-wise multiple comparisons were done with Least Significant Difference, using SPSS statistical software (SPSS, Chicago, USA). P-values <0.05 were regarded as statistically significant.
Results
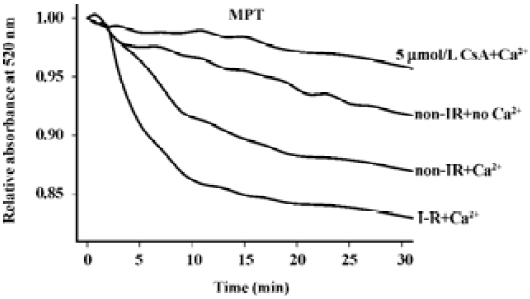
Ca2+, when added at a final concentration of 1 µmol/L in the presence of PO43–, enhanced the time-dependent decrease in absorbance at 520 nm, an indirect measure of mitochondrial swelling, in a suspension of heart mitochondria (Figure 2). The Ca2+-stimulated mitochondrial swelling was largely inhibited by CsA (5 µmol/L). While mitochondria from non-I–R and I–R hearts exhibited a comparable and small extent of swelling in the absence of Ca2+, they showed a larger extent of swelling in the presence of Ca2+, with the extent of Ca2+-induced PT of I–R mitochondria being larger than that of the non-I–R control. The extent of CsA-inhibitable mitochondrial swelling induced by Ca2+ was computed and served as an indirect measure of mitochondrial PT, as described in Materials and methods.
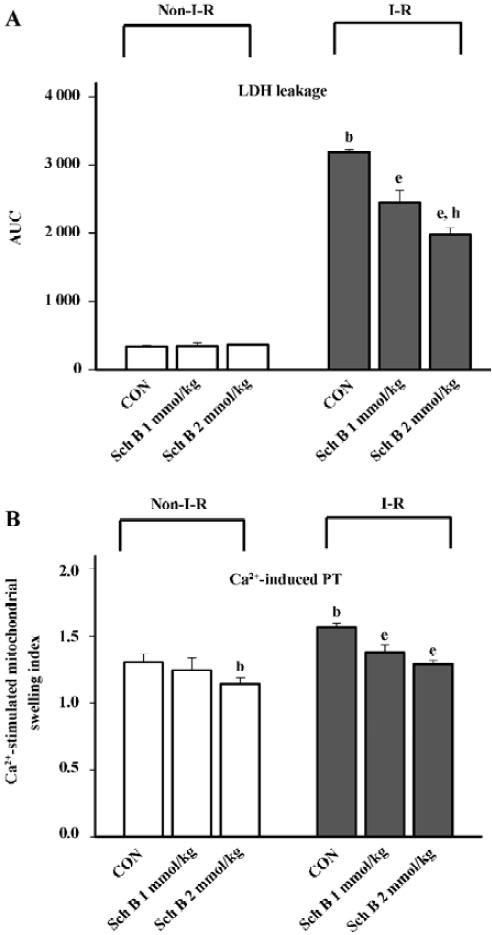
Pretreatment with Sch B (1–2 mmol/kg, po) protected against myocardial I–R injury in rats, as evidenced by the significant decrease in the extent of LDH leakage (23%–38%), when compared with the unpretreated control (Figure 3A). While Sch B treatment at the same dosage suppressed Ca2+-stimulated PT in heart mitochondria (7%–15%), when compared with the untreated control, I–R caused a slight, but significant increase in the sensitivity of heart mitochondria to Ca2+-stimulated PT (by 18%) in the rats (Figure 3B). The inhibitory effect of Sch B treatment on mitochondrial PT was also evident after the I–R challenge. The pretreatment with Sch B significantly reduced the extent of mitochondria PT (by 9%–18%) in I–R hearts, when compared with the unpretreated I–R control (Figure 3B).
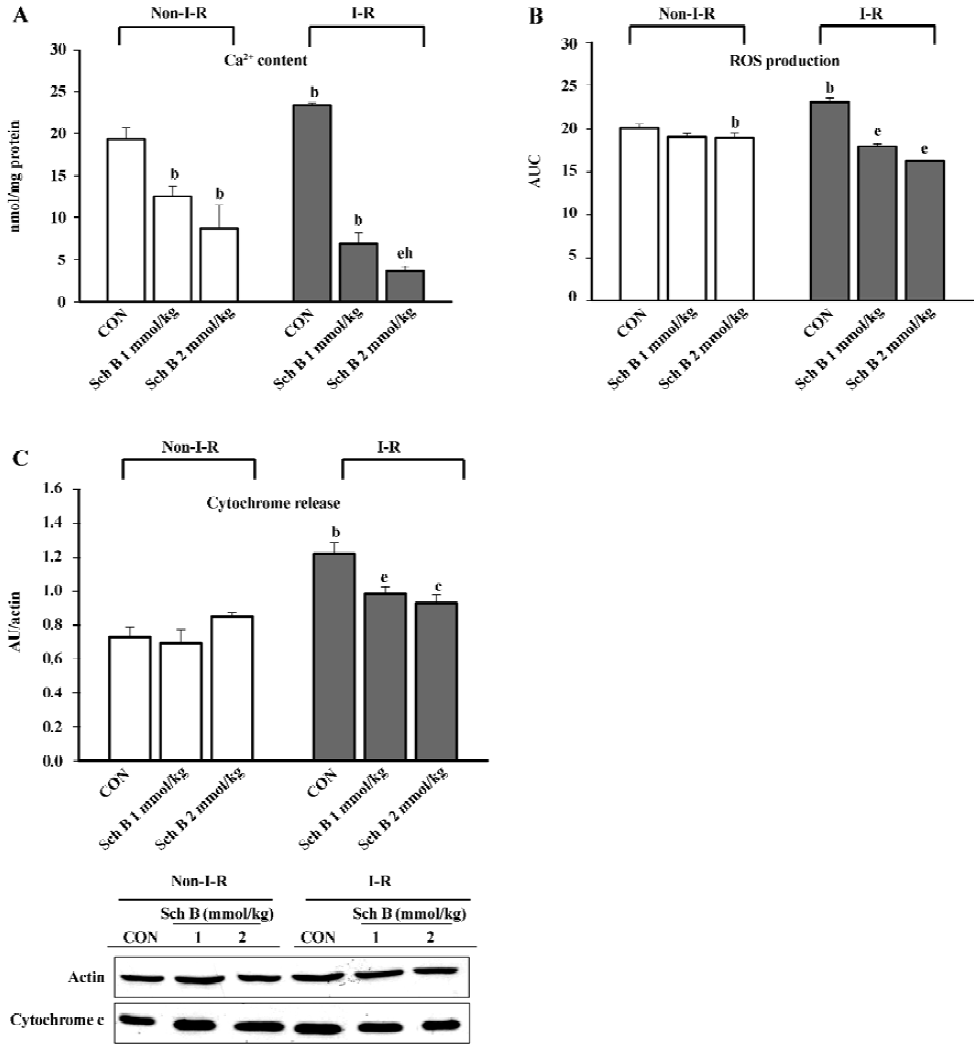
I–R caused significant increases in the mitochondrial Ca2+ level (21%), as well as the extent of mitochondrial ROS production (15%) and cytochrome c release (67%) the in rat hearts (Figure 4A–4C). While Sch B treatment dose-dependently decreased the mitochondrial Ca2+ level (by 35%–55%) in non-I–R rat hearts (Figure 4A), the extent of mitochondrial ROS production and cytochrome c release remained relatively unchanged (Figure 4B,4C). Sch B pretreatment dose-dependently suppressed the I–R-induced increases in the mitochondrial Ca2+ level, ROS production, and cytochrome c release to varying degrees (71%–85%, 23%–30%, and 19%–24%, respectively) when compared with the respective Sch B unpretreated control (Figure 4A–4C).
Discussion
Mitochondria not only generate ATP through oxidative phosphorylation, but are also an important source of ROS in mammalian cells under both physiological[21] and pathological conditions, such as myocardial I–R injury[22]. As far as the maintenance of cellular structural and functional integrity is concerned, the mitochondria serve as coordinators for cell survival and death[23]. In this regard, the opening of the mitochondrial transition pore is critically involved in the development of cellular dysfunction and cell death[24]. In the present study, the I–R-induced myocardial injury was associated with the increase in the sensitivity of the mitochondria to Ca2+-induced PT, as assessed by in vitro measurement of mitochondrial swelling. Core components of mitochondrial PT pore putatively consist of a voltage-dependent anion channel, an adenine nucleotide translocase (ANT), and cyclophilin D that displays peptidyl-prolyl cis-trans isomerase (PPIase) activity[7]. An excess amount of Ca2+ would compete over ATP and activate PPIase, resulting in a conformation change that converts the ANT complex into a non-specific pore[25]. The opening of mitochondrial PT pores, either in in vivo or in vitro conditions, is triggered by high mitochondrial Ca2+ content and other stimuli, including oxidants and the depletion of adenine nucleotides[26]. Consistent with this, mitochondria isolated from I–R rat hearts were found to have increased Ca2+ content as well as increased extent of ROS production. The disruption of structural integrity of mitochondria, as implicated by increases in the sensitivity to mitochondria PT and the extent of mitochondrial ROS production, from I–R myocardial tissues was evidenced by the increase in cytochrome c release in vivo. The cardioprotection afforded by Sch B pretreatment against I–R injury was associated with the improvement in mitochondrial integrity, as indicated by decreases in the sensitivity to Ca2+-induced PT, as well as the extent of ROS production and cytochrome c release. This is also corroborated by our recent findings that showed the preservation of mitochondrial ATP generation capacity by Sch B pretreatment in I–R rat hearts[3]. The leakage of cytochrome c in non-I–R hearts, as evidenced by the presence of cytosolic cytochrome c, is likely caused by the artifact arising from tissue homogenization, as was the case for Kim et al[27].
Cytosolic Ca2+ content increases during myocardial I–R[28], leading to the accumulation of Ca2+ in the mitochondria via the uptake by the inner membrane Ca2+ uniporter[29,30]. The mitochondrial Ca2+ overload not only generates energy-consuming futile cycles that divert the use of the inner membrane proton gradient to cation transport rather than ATP production[30], but also predisposes the mitochondria to PT[31]. The mitochondrial PT further jeopardizes the cellular energy status, and the consequent loss of ion homeostasis can lead to necrotic cell death. The release of cytochrome c from the mitochondrial inner membrane, which is believed to be an event secondary to the onset of mitochondrial PT[32], is a key step leading to apoptosis[11]. While under the present experimental conditions the relative contribution of necrotic and apoptotic cell death to I–R-induced tissue injury remains to be determined, the finding of decreased sensitivity of mitochondria isolated from Sch B-pretreated hearts to Ca2+-stimulated PT suggests that the increase in mitochondrial resistance to Ca2+-stimulated PT may play an important role in protecting against myocardial I–R injury. The observation that Sch B treatment decreased the mitochondrial Ca2+ level in both non-I–R and I–R hearts may be caused by the inhibition of the inner membrane Ca2+ uniporter and hence the Ca2+ uptake by Sch B. In this regard, Sch B was found to inhibit the P-glycoprotein, an ATP-dependent drug transporter in cancer cells[33].
Glutathione plays an important role in numerous cellular functions, including the regulation of Ca2+ homeostasis and detoxification of ROS[34,35]. Previous studies have demonstrated the ability of Sch B to enhance mitochondrial gluta-thione redox status and protect against I–R-induced myocardial injury[3]. The decrease in mitochondrial sensitivity to Ca2+-stimulated PT by Sch B, as observed in the present study, may be related to the enhancement of mitochondrial glutathione redox status. In this regard, 1 of the 2 voltage-sensitive sites of mitochondrial PT pore was found to be gated by a critical dithiol that was sensitive to glutathione redox status, and the oxidation of GSH could open the pore[36,37]. Furthermore, the modification of a specific thiol group on the ANT, either by oxidative stress or thiol reagents, also decreased adenine nucleotide binding and activated the PT pore[38]. Increased oxidative stress enhanced cyclophilin binding and hence increased the sensitivity of mitochondrial PT[38]. Consistently, the cardioprotection against I–R injury and the increase in the resistance of mitochondria to Ca2+-induced PT afforded by Sch B pretreatment were abolished by GSH depletion (unpublished data). The observation that a smaller degree of inhibition of Ca2+-induced mitochondrial PT than that of myocardial I–R injury afforded by Sch B treatment supports the notion that the decrease in the sensitivity of mitochondria to PT is an effect secondary to the enhancement of mitochondrial glutathione redox status. The direct effect of increasing the mitochondrial GSH level and/or the indirect effect on removing mitochondrial-derived ROS produced by Sch B treatment may probably increase the threshold for mitochondrial PT pore opening in the presence of Ca2+. It is unlikely that Sch B can produce a direct effect on mitochondrial PT pore opening because Sch B, when being supplemented in the perfusate, could not protect against I–R injury in isolated rat hearts[4].
In conclusion, I–R caused an increase in the sensitivity of myocardial mitochondria to Ca2+-stimulated PT in vitro. The cardioprotection afforded by Sch B pretreatment against I–R injury was paralleled by the decrease in the sensitivity of the myocardial mitochondria to Ca2+-stimulated PT, particularly under I–R conditions. The results suggest that the cardioprotection afforded by Sch B pretreatment against I–R injury may at least in part be attributed to the increase in the resistance of the myocardial mitochondria to Ca2+-stimulated PT.
References
- Ko RKM, Mak DHF. Schisandrin B and other dibenzooctadiene lignans. In: Packer L, Ong NO, Halliwell B, editors. Herbal and traditional medicine: molecular aspects of health. New York: Marcel Dekker; 2004. p 289–314.
- Yim TK, Ko KM. Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol Cell Biochem 1999;196:151-6.
- Chiu PY, Ko KM. Time-dependent enhancement in mitochondrial glutathione status and ATP generation capacity by schisandrin B treatment decreases the susceptibility of rat hearts to ischemia-reperfusion injury. Biofactors 2003;19:43-51.
- Ko KM, Yiu HY. Schisandrin B modulates the ischemia-reperfu-sion induced changes in non-enzymatic antioxidant levels in isolated-perfused rat hearts. Mol Cell Biochem 2001;220:141-7.
- Chiu PY, Ko KM. Schisandrin B protects myocardial ischemia-reperfusion injury partly by inducing Hsp25 and Hsp70 expression in rats. Mol Cell Biochem 2004;266:139-44.
- Honda HM, Ping P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc Drugs Ther 2006;20:425-32.
- Tsujimoto Y, Shimizu S. Role of mitochondrial membrane permeability transition in cell death. Apoptosis 2007;12:835-40.
- Al Nasser I, Crompton M. The reversible Ca2+-induced permea-bilization of rat liver mitochondria. Biochem J 1986;239:19-29.
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366:177-96.
- Redegeld FA, Moison RM, Koster AS, Noordhoek J. Depletion of ATP but not of GSH affects viability of rat hepatocytes. Eur J Pharmacol 1992;228:229-36.
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309-12.
- Kang PM, Haunstetter A, Aoki H, Usheva A, Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res 2000;87:118-25.
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 1999;79:1127-55.
- Fontaine E, Bernardi P. Progress on the mitochondrial permeability transition pore: regulation by complex I and ubiquinone analogs. J Bioenerg Biomembr 1999;31:335-45.
- Ip SP, Poon MKT, Wu SS, Che CT, Ng KH, Kong YC, et al. Effect of schisandrin B on hepatic glutathione antioxidant system in mice: protection against carbon tetrachloride toxicity. Planta Med 1995;61:398-401.
- Li PC, Mak DHF, Poon MKT, Ip SP, Ko KM. Myocardial protective effect of Sheng Mai San (SMS) and a lignan-enriched extract of Fructus Schisandrae, in vivo and ex vivo. Phytomedicine 1996;III:161-5.
- Chiu PY, Tang MH, Mak DHF, Poon MKT, Ko KM. Hepato-protective mechanism of schisandrin B: Role of mitochondrial glutathione antioxidant status and heat shock proteins. Free Radic Biol Med 2003;35:368-80.
- Leung HY, Chiu PY, Poon MKT, Ko KM. A Yang-invigorating Chinese herbal formula enhances mitochondrial functional ability and antioxidant capacity in various tissues of male and female rats. Rejuven Res 2005;8:238-47.
- Kowaltowski AJ, Netto LES, Vercesi AE. The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. J Biol Chem 1998; 273: 12 766–9.
- Menze MA, Hutichsin K, Laborde SM, Hoard SC. Mitochondrial permeability transition in the Crustacean Artemia franciscana: absence of a calcium-regulated pore in the face of profound calcium storage. Am J Physiol Regul Integr Comp Physiol 2005;289:R68-76.
- Demin OV, Kholodenko BN, Skulachev VP. A model of O2.-generation in the complex III of the electron transport chain. Mol Cell Biochem 1998;184:21-33.
- Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, . Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 1993; 268: 18 532–41.
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998;60:619-42.
- Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic Biol Med 2005;38:687-97.
- Galat A, Metcalfe SM. Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol 1995;63:67-118.
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:C817-33.
- Kim GT, Chun YS, Park JW, Kim MS. Role of apoptosis-inducing factor in myocardial cell death by ischemia–reperfusion. Biochem Biophys Res Commun 2003;309:619-24.
- Ataka K, Chen D, Levitsky S, Jimenez E, Feinberg H. Effect of aging on intracellular Ca2+, pHi, and contractility during ischemia and reperfusion. Circulation 1992;86:II371-6.
- Grover G.J, Dzwonczyk S, Sleph PG. Ruthenium red improves postischemic contractile function in isolated rat hearts. J Cardio-vasc Pharmacol 1990;16:783-9.
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol 1990;258:C755-86.
- Kushnareva YE, Sokolove PM. Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane. Arch Biochem Biophys 2000;376:377-88.
- Crompton M, Costi AA. Heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J 1990;266:33-9.
- Pan Q, Wang T, Lu Q, Hu X. Schisandrin B–a novel inhibitor of P-glycoprotein. Biochem Biophys Res Commun 2005;235:406-11.
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem 1983;52:711-60.
- Reed DJ. Glutathione: toxicological implications. Annu Rev Pharmacol Toxicol 1990;30:603-31.
- Costantini P, Petronilli V, Colonna R, Bernardi P. On the effects of paraquat on isolated mitochondria. Evidence that paraquat causes opening of the cyclosporin A-sensitive permeability transition pore synergistically with nitric oxide. Toxicology 1995;99:77-88.
- Reed DJ, Savage MK. Influence of metabolic inhibitors on mitochondrial permeability transition and glutathione status. Biochim Biophys Acta 1995;1271:43-50.
- Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cycloporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects heart from ischaemia/reperfusion injury. Mol Cell Biochem 1997;174:167-72.