
NF-κB downregulation may be involved the depression of tumor cell proliferation mediated by human mesenchymal stem cells1
Introduction
Emerging evidence has shown that hematopoietic stem cell transplantation can have tumor inhibitory effects in patients with solid tumors, such as a case of regression of unresectable, large pancreatic tumor and a case of attenuation of multiple metastases of fibroblastic osteosarcoma following non-myeloablative stem cell transplantation[1–3]. Adult human and murine hematopoietic stem cells and progenitor cells display a tropism for intracerebral gliomas in mice[4]. Likewise, experiments show that human neural stem cells can inhibit mouse glioma growth[5]. Mesenchymal stem cells (MSCs) have been identified as a kind of cell with differentiation capacity along mesodermal lineages, migrate and activate at a site of disease, and could be employed in cell-replacement therapy and gene therapy[6–8]. Human MSCs (hMSCs) can target microscopic tumors and contribute to stem cell-based anticancer gene therapeutic approaches. Animal studies have demonstrated that gene-modified hMSC may serve as a platform for delivery of biological agents in antitumors[9]. Human bone marrow-derived MSC may have a tropism for brain tumors through the blood and increase animal survival. Moreover, hMSC were recruited exclusively into the brain glioma contrasted with the widespread distribution of fibroblasts without tumor specificity in vivo[10]. However, according to the recent studies, there are discrepancies about the effect of standard MSCs on tumor cells. It is described that unmodified MSCs are able to home to several different tumor cells in mice and enhance their growth[11]. Other studies suggest that unmodified hMSCs can inhibit the growth of tumor cells, for example, the homing of hMSCs to sites of Kaposi’s sarcoma and potently inhibit tumor growth in vivo, in the meantime downregulating the Akt protein kinase of some types of tumor cells by cell–cell direct contact[12]. The opposite of the results on the intrinsic effects of MSC on tumor growth needs further examination. Moreover, stem cells also show an antitumor effect in metastatic cancers from breast, kidney, ovaries, prostate, and pancreas[13]. Importantly, a study indicates that a product containing stem cell differentiation stage factors inhibit hepatocellular carcinoma growth in vivo and in vitro[14]. In our previous study, we revealed that hMSCs are able to inhibit the proliferation of breast cancer cells via the Wnt/β-catenin pathway[15]. Taken together, a number of reports show that stem cells display intrinsic antitumor effects, especially stem cells home to tumorigenesis, indicating a particular utility for tumor. However, the underlying molecular mechanism that may be involved in multiple signaling pathways remains unclear.
NF-κB is present broadly in the majority of cells and is a key transcription factor, which is involved in cellular proliferation, differentiation, carcinogenesis, and apoptosis. The family of NF-κB transcription factors include NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel. The activation of NF-κB has been implicated in human prostate cancer and tongue carcinoma cells[16–18]. The upregulation of the NF-κB signaling pathway in human thyroid cancer cells leads to apoptotic resistance and promotes invasive ability, which is significantly inhibited by a specific NF-κB inhibitor[19]. The inhibition of NF-κB activation leads to the downregulation of gene products involved in cell survival, proliferation, and invasion[20]. NF-κB significantly inhibits cell growth and promotes cell death in human hepatocellular cancer cells and ovarian cancer cells[21–23]. Oncogenic H-Ras, an upstream activator of Akt, requires NF-κB to suppress transformation-associated apoptosis[24]. Deguelin, an Akt inhibitor, suppresses NF-κB leading to the potentiation of apoptosis and the inhibition of cellular invasion[25]. Embelin, an inhibitor of XIAP blocks the NF-κB signaling pathway and leads to the suppression of NF-κB-regulated anti-apoptotic and metastatic gene expression[26]. Overall, the inhibition of NF-κB activation provides convincing evidence for the critical role of antitumor therapy.
We previously found that Dkk-1 released from hMSCs contributed to the depression of the proliferation of breast cancer cells via the Wnt/β-catenin pathway[15]. Moreover, in the present study, we investigated the role of NF-κB in the depression of tumor cells mediated by hMSCs. Our finding showed that NF-κB downregulation may be involved in the inhibition of tumor cells mediated by hMSCs.
Material and methods
Cell culture The hMSCs designated Z3 were established from human dermis tissues of a fetus aborted at 4 months’ gestation, and immortalized by the stable transfection of a pGRN145 plasmid containing the cloned hTERT gene[27]. The FACScan flow cytometer showed that hTERT(+) cells were positive for CD29, CD44, CD105, and CD166, while CD31, CD45, CD34, vWF, and HLA-DR were negative. Under suitable conditions, hTERT(+) cells have the ability of multiple lineage differentiation, including bone, fat, and nerve. The hMSC-designated BMMS-03, derived from human fetal bone marrow at 4 months, were established without immortalization[28]. The FACScan flow cytometer showed that BMMS-03 cells were positive for CD105 and CD166, while CD34 was negative. For the experiments, Z3 and BMMS-03 were cultured in Dulbecco’s modified Eagle’s medium with low glucose and Iscove’s modified Dulbecco’s medium at 1:1 supplemented with 10% fetal calf serum (Gibco, USA), H7402/HepG2 human hepatoma cells and MCF-7 human breast cancer cells (low metastasis potential)/LM-MCF-7 (high metastasis potential, a subclone derived from lung metastasis MCF-7 cells) human breast cancer cells were cultured in RPMI-1640 containing 10% fetal calf serum[29]. The cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C.
Cell treatment Z3 or BMMS-03 cells were cultured as described above 100% confluence. The supernatant derived from the Z3 or BMMS-03 cultures was harvested and stored at –80 °C until use as conditioned media for cell treatment. Synchronized H7402/HepG2 human hepatoma cells or MCF-7/LM-MCF-7 human breast cancer cells were pretreated with a mixture of RPMI-1640 containing 10% fetal calf serum and Z3- or BMMS-03-conditioned media (199:1, 49:1, 9:1, 4:1, or 1:1, namely 0.5%, 2.0%, 10%, 20%, or 50%) for 48–96 h, respectively. During that time, the culture media were replaced every 24 h. The treated tumor cells were examined by the 5-bromodeoxyuridine (BrdU) incorporation assay, flow cytometry assay, reporter gene assay, real-time PCR, and Western blot analysis, respectively.
BrdU incorporation assay DNA synthesis or cell proliferation was measured by the BrdU incorporation assay. BrdU (Sigma, USA) was used to incorporate into DNA in place of thymindine[30]. Briefly, the H7402 cells or MCF-7 cells were seeded in a 96-well plate, respectively, and incubated with treatment of 50% hMSC conditioned media for 48 h. BrdU (10 µmol/L final concentration) was added and the cells were re-incubated for an additional 4 h at 37 °C. The cells, which incorporated BrdU into DNA, were detected using a monoclonal antibody against BrdU (1:300 dilution, Neomarkers, USA) at room temperature for 2 h. A goat anti-mouse immunoglobulin G-labeled fluorescein-isothiocyanate (1:100 dilution; Sigma, USA) was used, as was propidium iodide (Sigma, USA) nuclear counterstaining. The results were expressed as the percentage of BrdU-positive cells over the propidium iodide-positive cells.
Flow cytometry analysis Proliferation was assessed by flow cytometry analysis. MCF-7 cells treated with 20% or 50% Z3- or BMMS-03-conditioned media as above for 48 h were used for the flow cytometry analysis. The cells were suspended in phosphate-buffered saline after being fixed with ice-cold 70% alcohol and were stained with propidium iodide (50 µg/mL). The proliferation index (PI) was determined from the flow cytometry analysis data according to the following formula: PI=(G2/M+S)÷(G0/G1+S+G2/M)×100%[31].
Transfection and reporter gene assay The activity of NF-κB was determined by transfecting the cells with a target gene-dependent reporter plasmid and measuring luciferase activity. The pGL-NF-κB reporter plasmid containing the NF-κB-dependent luciferase reporter gene (provided by Dr Chuan-shu Huang form New York University, New York, USA) and the pRL-TK plasmid containing the renilla luciferase gene as an internal control were cotransfected into MCF-7/LM-MCF-7 breast cancer cells and H7402/HepG2 human hepatoma cells in triplicate by using Lipofectamine 2000 (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s recommendation, when treated with conditioned media of Z3 cells or BMMS-03 cells.The cells were harvested 48 h after transfection, and the activities of target gene luciferase and renilla luciferase were determined with the dual luciferase reporter assay kit (Promega, San Luis Obispo, CA, USA). Luciferase and renilla luciferase luminiscence were measured by using a luminometer. All of the data shown in this study were obtained from at least three independent experiments.
Quantitative real-time PCR Quantitative real-time PCR was performed to examine the expression level of NF-κB in treated cells and untreated cells by a LightCycler PCR analyzer (Roche, Mannheim, Germany) using the one-step RT-PCR system. Relative quantitation was performed using the LightCycler fast start DNA master SYBR green I kit (Roche, Germany) to detect PCR products in real time with the LightCycler. The oligonucleotide primers for PCR were based on published mRNA sequences as follows: human NF-κB2 sense primer, 5'-CATGGAGAGTTGCTACAACC-3'; human NF-κB2 antisense primer, 5´-TCTCTGCTTAGGCTGTTCCA-3'; human GAPDH sense primer, 5'-TGTTGCCATCAATGACCCCTT-3'; and GAPDH antisense primer, 5'-CTCCACGACGTACTCAGCG-3'. After denaturation at 95 °C for 10 min, PCR was performed for 45 cycles (10 s at 95 °C, 5 s at 60 °C, and 15 s at 72 °C). Each experiment was repeated 5 times independently. All amplified products were sequence verified. Control reactions were performed in the absence of reverse transcriptase and were negative. The target gene transcripts relative to the housekeeping gene GAPDH were quantified, and the results were expressed as a relative value compared with GAPDH.
Western blot analysis The expression levels of NF-κB and the phosphorylation of inhibitor κBα(p-IκBα) were detected in H7402 cells and MCF-7 cells by Western blot analysis. After treatment with 10% conditioned media of Z3 or BMMS-03 cells for 96 h, the MCF-7 cells and H7402 cells were lysed in lysis buffer (62.5 mmol/L Tris-HCl, pH 6.8, 2% SDS, 5% 2-mercaptoethanol, and 10% glycerol) at 4 °C for 20 min. The lysate was centrifuged at 10 000×g at 4 °C for 20 min. The protein concentrations of the supernatant were determined with a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). The proteins in the cell lysate were separated on a 12% SDS-PAGE gel and transferred to a nitrocellulose filter, then incubated with an antibody against NF-κB at a dilution of 1:400 (NeoMarkers, USA) or an antibody against p-IκBα at a dilution of 1:300 (Santa Cruz, CA, USA) for 2 h at room temperature, respectively, and re-incubated with a peroxidase-conjugated secondary antibody for 1 h at room temperature. The proteins of NF-κB and p-IκBα were detected by the enhanced chemiluminescence system (Sigma, USA).
Statistical analysis The analysis of data was carried out with Student’s t-test.
Results
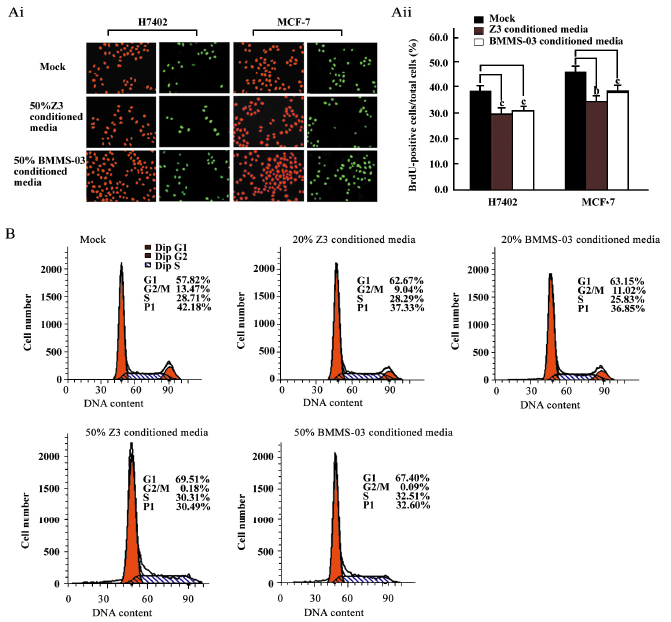
Depression of the proliferation of tumor cells treated with conditioned media from hMSC We investigated the effect of hMSCs on tumor cells by using conditioned media from hMSC culture. The proliferation of H7402 and MCF-7 cells treated with 20% or 50% conditioned media from Z3 cell or BMMS-03 cell cultures were assessed by the BrdU incorporation assay and flow cytometry analysis. The BrdU incorporation assay indicated that the positive cells in the S phase of the treated group were significantly lower than that in the control group (Figure 1A). The average positive rate of BrdU staining was 38.2% in the control group of H7402 cells; however, the average positive rate of BrdU staining was 29.2% in the H7402 cells inoculated with 50% Z3 conditioned media (P<0.01 vs control), and was 30.1% in the H7402 cells inoculated with 50% BMMS-03-conditioned media (P<0.01 vs control). A similar result was obtained in the MCF-7 cells. The average positive rate of BrdU staining was 46.3% in the control group of MCF-7 cells; however, the average positive rate of BrdU staining was 33.7% in the cells inoculated with 50% Z3 conditioned media (P<0.05 vs control), and was 37.2% in the cells inoculated with 50% BMMS-03-conditioned media (P<0.01 vs control). Furthermore, the flow cytometry analysis was carried out in MCF-7 cells treated with conditioned media from hMSCs at 2 concentrations of 20% and 50% for 48 h. The flow cytometry analysis showed that the PI decreased in the treated cells compared with the control. The PI was 37.33% or 36.85% in MCF-7 cells treated with 20% Z3- or BMMS-03- conditioned media, and 30.49% or 32.60% treated with 50% conditioned media from Z3 or BMMS-03, but the PI was 42.18% in the control cells (P<0.01; Figure 1B). The data indicate that the conditioned media from hMSCs are able to inhibit the proliferation of hepatoma cells and breast cancer cells.
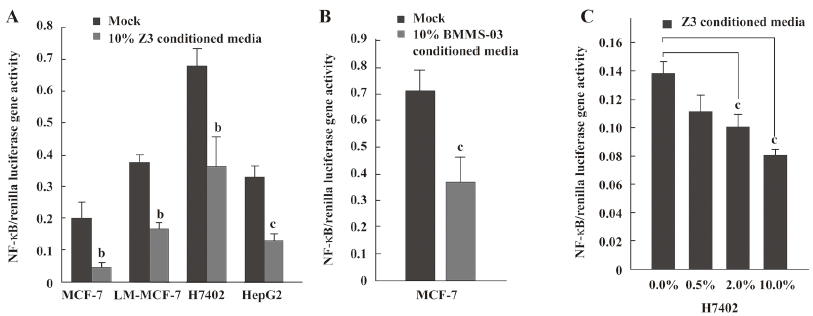
NF-κB downregulation may involve the depression of tumor cells mediated by conditioned media from hMSCs Next, we sought to investigate the molecular mechanisms underlying the inhibitory effect of hMSCs on tumor cells. We addressed whether or not NF-κB was involved in the depression of tumor cells mediated by conditioned media from hMSCs. First, we examined the transcriptional activity of NF-κB in tumor cells after the treatment with conditioned media from hMSCs. The NF-κB-dependent luciferase plasmid was transfected into MCF-7/LM-MCF-7 breast cancer cells and H7402/HepG2 hepatoma cells in triplicate when treated with 10% conditioned media from Z3 cells. It was found that the activity of NF-κB-dependent luciferase was downregulated (Figure 2A). A similar result was obtained in MCF-7 human breast cells employing 10% conditioned media from BMMS-03 cells (Figure 2B). The treatment of MCF-7 cells with conditioned media from Z3 cells of 0.5%, 2.0%, or 10.0% significantly resulted in the depression of NF-κB in a dose-dependent fashion (Figure 2C), suggesting that the NF-κB-mediated transactivation of luciferase activity in tumor cells was downregulated by conditioned media from hMSCs.
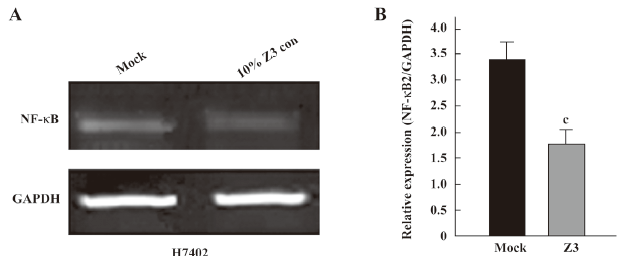
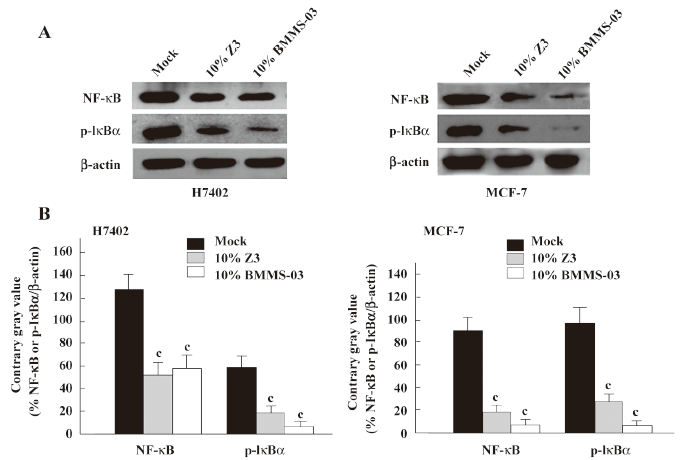
Second, we examined the expression of NF-κB at the mRNA and protein levels. The NF-κB2 and GAPDH mRNA levels were determined by quantitative real-time PCR. The PCR products for NF-κB2 and GAPDH were 131 and 202 bp, respectively. The results showed that the expression level of NF-κB2 mRNA was downregulated in H7402 cells treated with 10% Z3 conditioned media (Figure 3). The Western blot analysis revealed that the expression levels of NF-κB were downregulated in H7402 and MCF-7 cells treated with 10% conditioned media from Z3 or BMMS-03 stem cells, and the downregulation of NF-κB in treated cells was accompanied by low p-IκBα (Figure 4). Taken together, our data indicate that some soluble factors in the conditioned media from Z3 and BMMS-03 cells were responsible for the suppression of hepatoma cells and breast cancer cells, in which NF-κB downregulation may involve in the inhibition.
Discussion
With the development of study in stem cell biology, more and more attention has been paid to the research of the relationship between stem cells and tumor cells. However, to date, the molecular mechanism of the inhibitory effect of stem cells on the phenotype of tumor cells remains unclear. Stem cells share several characteristics of cancer cells, including loss of contact inhibition and immortality. Importantly, stem cells and tumor cells have similar signaling pathways that regulate self-renewal and differentiation, such as Wnt, BMP, mitogen-activated protein kinase (MAPK), and Notch pathways, which determine the diverse developmental fates of cells[32–34]. The microenvironment around stem cells plays an essential role in preventing carcinogenesis by providing primarily inhibitory signals for both proliferation and differentiation[35]. Understanding the signaling circuitry involved in cancer cells may provide an insight into the molecular mechanisms of tumorigenesis. Some researchers have observed that hMSCs are able to home to the sites of tumorigenesis, and the tropism of MSC may possibly be suitable for cancer therapy.
In the present study, we treated human hepatoma cells and human breast cancer cells with conditioned media from Z3 or BMMS-03 hMSCs. The BrdU incorporation assay and flow cytometry assay demonstrated that the proliferation ability of H7402 and MCF-7 cells was inhibited significantly with treatment of 20% or 50% conditioned media from Z3 or BMMS-03 hMSCs. These findings strongly suggest that some soluble factors in the conditioned media from Z3 or BMMS-03 cells are able to inhibit tumor cells. We hypothesize that the interaction of tumor cells and hMSCs is influenced by ligand-dependent extracellular signaling, in which NF-κB may be involved in the signalings. Therefore, we investigated the regulation of NF-κB at the levels of transcription, mRNA, protein and activity regulation. The results demonstrated that the transcriptional activity of NF-κB was downregulated in H7402/HepG2 human hepatoma cells and MCF-7/LM-MCF-7 human breast cancer cells after treatment with 10% conditioned media from Z3 or BMMS-03 cells by measuring luciferase activity. Real-time PCR and the Western blot analysis showed the downregulation of NF-κB at the mRNA and protein levels. In the resting state, the activity of NF-κB is sequestered in the cytoplasm as an inactive precursor complex with inhibitory κB (IκB). Members of the IκB family include IκBα, IκBβ, IκBγ, IκBε, and Bcl-3. The biological activity of NF-κB is tightly regulated by IκB specific to IκBα[36]. It appears that the newly synthesized protein IκB rapidly re-associates with the newly released NF-κB, thereby markedly reducing the amount of NF-κB translocated into the nucleus for the activation of cytokine genes. The activity of the NF-κB transcription factor is regulated by p-IκB through multiple intracellular signal transduction pathways. Inflammatory cytokines, such as tumor necrosis factor (TNF), activate the IκB kinase (IKK) complex to phosphorylate the conserved N-terminal region of IκB proteins. Additionally, protein kinase C directly regulates the phosphorylation of IκB, which results in the release of NF-κB and the appearance of NF-κB DNA-binding activity[37]. Mitogen-activated/extracellular response kinase kinase 1 has been found to participate in IKK activation in response to TNF-β in 293 cells[38]. Once IκB is phosphorylated and degraded, active NF-κB is released and translocates into the nucleus to enhance the expression of genes. Further study was performed to determine the implications of this phenomenon with regard to p-IκB. Our results indicated that the downregulation of NF-κB and p-IκBα in treated cells was cooperatively regulated in H7402 and MCF-7 cells. A number of reports showed that NF-κB is regulated by the intracellular signal transduction cascades. A variety of active factors regulate the activity of NF-κB, such as TNF-α/β, interferonβ/γ, interleukin (IL)-1α, IL-1β, IL-2, TGF-β, and macrophage colony-stimulating factor. However, the factors in the conditioned media released from hMSCs that are responsible for the downregulation of NF-κB in tumor cells remain unclear.
Taken together, our study illustrates that conditioned media of hMSCs inhibits the proliferation of hepatoma cells and breast cancer cells, which may be related to multiple signaling pathways, such as the downregulation of either Wnt/β-catenin[18] and/or NF-κB, and/or others. The mechanism needs to be further investigated. The application of hMSCs may be used in the therapeutic strategy of cancers.
Acknowledgements
We thank Dr Chuan-shu HUANG (from Nelson Institute of Environmental Medicine, New York University School of Medicine, New York, USA) for providing the pGL-NF-κB and pRL-TK reporter plasmid.
References
- Omuro Y, Matsumoto G, Sasaki T, Tanaka Y, Maeda Y, Sakamaki H, et al. Regression of an unresectable pancreatic tumor following nonmyeloablative allogeneic peripheral-blood stem-cell transplantation. Bone Marrow Transplant 2003;31:943-5.
- Hentschke P, Barkholt L, Uzunel M, Mattsson J, Wersall P, Pisa P, et al. Low-intensity conditioning and hematopoietic stem cell transplantation in patients with renal and colon carcinoma. Bone Marrow Transplant 2003;31:253-61.
- Renga M, Pedrazzoli P, Siena S. Present results and perspectives of allogeneic non-myeloablative hematopoietic stem cell transplantation for treatment of human solid tumors. Ann Oncol 2003;14:1177-84.
- Tabatabai G, Bahr O, Mohle R, Eyupoglu IY, Boehmler AM, Wischhusen J, et al. Lessons from the bone marrow: how malignant glioma cells attract adult haematopoietic progenitor cells. Brain 2005;128:2200-11.
- Kim SK, Cargioli TG, Machluf M, Yang W, Sun Y, Al-Hashem R, et al. PEX-Producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin Cancer Res 2005;11:5965-70.
- Yuan X, Hu J, Belladonna ML, Black KL, Yu JS. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer Res 2006;66:2630-8.
- Wulf GG, Chapuy B, Trumper L. Mesenchymal stem cells from bone marrow. Phenotype, aspects of biology, and clinical perspectives. Med Klin (Munich) 2006;101:408-13.
- Chen XC, Wang R, Zhao X, Wei YQ, Hu M, Wang YS, et al. Prophylaxis against carcinogenesis in three kinds of unestablished tumor models via IL12-gene-engineered MSCs. Carcinogenesis 2006;27:2434-41.
- Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow–derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 2005;65:3307-18.
- Hung SC, Deng WP, Yang WK, Liu RS, Lee CC, Su TC, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res 2005;11:7749-56.
- Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst 2004;96:1593-603.
- Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med 2006;203:1235-47.
- Ringden O, Le Blanc K. Allogeneic hematopoietic stem cell transplantation: state of the art and new perspectives. APMIS 2005;113:813-30.
- Livraghi T, Meloni F, Frosi A, Lazzaroni S, Bizzarri TM, Frati L, et al. Treatment with stem cell differentiation stage factors of intermediate-advanced hepatocellular carcinoma: an open randomized clinical trial. Oncol Res 2005;15:399-408.
- Qiao L, Xu Z, Tan X, Ye L, Zhang X. Investigation of inhibition of Wnt/β-catenin pathway in breast cancer cells mediated by Dkk-1 released from human mesenchymal stem cells. Prog Biochem Biophys 2007;34:702-8.
- Uzzo RG, Crispen PL, Golovine K, Makhov P, Horwitz EM, Kolenko VM. Diverse effects of zinc on NF-κB and AP-1 transcription factors: implications for prostate cancer progression. Carcinogenesis 2006;27:1980-90.
- Karin M. Nuclear factor-κB in cancer development and progression. Nature 2006;441:431-6.
- Yao J, Duan L, Fan M, Wu X. NF-κB signaling pathway is involved in growth inhibition, G2/M arrest and apoptosis induced by trichostatin A in human tongue carcinoma cells. Pharmacol Res 2006;54:406-13.
- Palona I, Namba H, Mitsutake N, Starenki D, Podtcheko A, Sedliarou I, et al. BRAFV600E promotes invasiveness of thyroid cancer cells through NF-κB activation. Endocrinology 2006;147:5699-705.
- Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest 2001;107:241-6.
- Kokura S, Yoshida N, Sakamoto N, Ishikawa T, Takagi T, Higashihara H, et al. The radical scavenger edaravone enhances the anti-tumor effects of CPT-11 in murine colon cancer by increasing apoptosis via inhibition of NF-κB. Cancer Lett 2005;229:223-33.
- Poma P, Notarbartolo M, Labbozzetta M, Sanguedolce R, Alaimo A, Carina V, et al. Antitumor effects of the novel NF-κB inhibitor dehydroxymethyl-epoxyquinomicin on human hepatic cancer cells: analysis of synergy with cisplatin and of possible correlation with inhibition of pro-survival genes and IL-6 production. Int J Oncol 2006;28:923-30.
- Luo JL, Kamata H, Karin M. IKK/NF-κB signaling: balancing life and death––a new approach to cancer therapy. J Clin Invest 2005;115:2625-32.
- Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Mol Cell Biol 2000;20:1626-38.
- Nair AS, Shishodia S, Ahn KS, Kunnumakkara AB, Sethi G, Aggarwal BB. Deguelin, an Akt inhibitor, suppresses IκBα kinase activation leading to suppression of NF-κB-regulated gene expression, potentiation of apoptosis, and inhibition of cellular invasion. J Immunol 2006;177:5612-22.
- Ahn KS, Sethi G, Aggarwal BB. Embelin, an inhibitor of XIAP, blocks nuclear factor-κB (NF-κB) signaling pathway leading to suppression of NF-κB-regulated anti-apoptotic and metastatic gene products. Mol Pharmacol 2007;71:209-19.
- Zhao Z, Liao L, Cao Y, Jiang X, Zhao RC. Establishment and properties of fetal dermis-derived mesenchymal stem cell lines: plasticity in vitro and hematopoietic protection in vivo. Bone Marrow Transplant 2005;36:355-65.
- Zhang XD, Xu ZL, Ye LH, Dong N, Zhao CH, Cai B, et al. Isolation and identification of mesenchymal stem cell derived from human fetal bone marrow. Acta Scientiarum Naturalium Universitatis Nankaiensis 2006;39:107-10.
- Ye LH, Wu LY, Guo W, Ma HT, Zhang X. Screening of a sub-clone of human breast cancer cells with high metastasis potential. Zhonghua Yixue Zazhi 2006;86:61-5.
- Lengronne A, Pasero P, Bensimon A, Schwob E. Monitoring S phase progression globally and locally using BrdU incorporation in TK+ yeast strains. Nucleic Acids Res 2001;29:1433-42.
- Plas E, Carroll VA, Jilch R, Simak R, Mihaly J, Melchior S, et al. Variations of components of the plasminogen activation system with the cell cycle in benign prostate tissue and prostate cancer. Cytometry 2001;46:184-9.
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005;434:843-50.
- Varga AC, Wrana JL. The disparate role of BMP in stem cell biology. Oncogene 2005;24:5713-21.
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006;442:823-6.
- Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res 2006;66:4553-7.
- Huang TT, Miyamoto S. Postrepression activation of NF-κB requires the amino-terminal nuclear export signal specific to IκBα. Mol Cell Biol 2001;21:4737-47.
- La Porta CA, Comolli R. PKC-dependent modulation of IκBα-NF-κB pathway in low metastatic B16F1 murine melanoma cells and in highly metastatic BL6 cells. Anticancer Res 1998;18:2591-7.
- Mogensen TH, Melchjorsen J, Höllsberg P, Paludan SR. Activation of NF-κB in virus-infected macrophages is dependent on mitochondrial oxidative stress and intracellular calcium: downstream involvement of the kinases TGF-β-activated kinase 1, mitogen-activated kinase/extracellular signal-regulated kinase kinase 1, and Iκβ kinase. J Immunol 2003;170:6224-33.