
Antitumor activity of a novel bis-aziridinylnaphthoquinone (AZ4) mediating cell cycle arrest and apoptosis in non-small cell lung cancer cell line NCI-H4601
Introduction
Despite advances in cancer treatment in the past 2 decades, the prognosis of lung cancer patients has improved minimally. Lung cancer is the leading cancer killer in both men and women. There was an estimated 171 900 new cases of lung cancer and an estimated 157 200 deaths from lung cancer in the Unite States in 2003[1]. There are 2 types of lung cancer cells, small cell lung cancer and non-small cell lung cancer. Non-small cell lung cancer (NSCLC) heterogeneously aggregates at least 3 distinct histological lung cancers, including epidermoid or squamous carcinoma, adenocarcinoma, and large cell carcinoma. NSCLC is the most common histological cell type of all lung cancers, and there is no curative treatment available for the advanced stages of these diseases. Although chemotherapy is an esta-blished treatment for advance NSCLC, it offers only a limited survival benefit at the expense of substantial toxicity, drug resistance, and poor target specificity. Thus, there is a need for innovative strategies that effectively treat patients and exhibit more favorable safety profiles in advanced NSCLC.
The ideal bioreductive drug should be administered as an inactive prodrug that is only activated under low-oxygen conditions by 1 or 2 electron reductases[2–4]. Aziridine-substituted benzoquinone, such as mitomycin C, RH1, and tirapazamine (TPZ), are 3 principal aziridinyl quinone classes of hypoxia-specific cytotoxins that are being developed for clinical use[5–7]. In the case of di-aziridinyl-substituted quinone, this highly cytotoxic, bifunctional, alkylating agent can cross-link DNA in cells which results in inducing complicated cellular mechanisms leading to cell death by apoptosis such as TPZ and CI-1010 or necrosis[8,9].
Tumor tissue had lower oxidative reduction (redox) potential relative to most normal tissues which could increase the reductive activation of these quinone derivatives in tumors[2]. Therefore, the selectivity of bioreductive drugs is governed not only by the difference in oxygen tension between tumors and normal tissues, but also by levels of enzymes catalyzing bioreductive activation such as DT-diaphorase[3,10–11]. This fact led to the 1990 publication of the concept of “enzyme-directed bioreductive development” by Workman and Walton[12].
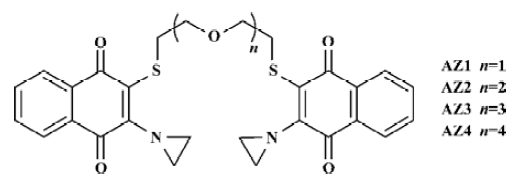
In this study, one of these bis-aziridinylnaphthoquinone analogues, designated as AZ4 (Figure 1) was found to inhibit NSCLC cell growth in vitro. The mechanism of AZ4 to the NSCLC cell H460 was investigated, including cell cycle arrest and apoptosis.
Materials and methods
Synthesis of AZ1 to AZ4 AZ1 to AZ4 were prepared by following our previously published methods[13]. Stock solutions (10 mmol/L) were freshly prepared in DMSO (Sigma, St Louis, MO, USA) and the series was diluted and directly added into the cultured cells from 20 µmol/L to 0.2 µmol/L.
Cell culture The cell line H460 (human NSCLC) was cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS), 2 mmol/L L-glutamine, 25 mmol/L hydroxyethyl-1-piperazine-ethanesulfonic acid (HEPES). The normal cell MRC-5 (human fetal lung fibroblast cell line) was cultured in Dulbecco’s modified eagle’s medium (DMEM) with 10% FBS, 2 mmol/L L-glutamine, and miminal essential medium (MEM) non-essential amino acid. The cell culture medium for those two cell lines all contained penicillin-streptomycin and fungizone. All the medium and supplements were purchased from Gibco Laboratories (Grand Island, NY, USA). All of the cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C. The amount of cells was counted after trypsinization by a Neubauer hemocytometer (VWR, Scientific Corp, Philadelphia, PA, USA).
Cytotoxicity assay (MTT assay) The MTT assay was according to the method of Skehan et al[14]. Two cell lines were seeded in 96-well flat-bottomed microtiter plates (3000–5000 cells/well), respectively. The cells were incubated for 24 h with drugs, applied as serial 1:2 dilutions (100 µL/well) ranging from 20 µmol/L down to 0.2 µmol/L or 0.1% DMSO as control. Twenty microliters of MTT (5 mg/mL) was added to each well and incubated for 4 h at 37 °C. The formazan product was dissolved by adding 100 µL DMSO to each well, and the plates were read at 550 nm. All measurements were performed in triplicate and each experiment was repeated at least 3 times. The IC50 was calculated from the 50% formazan formation compared with the control without the addition of drugs.
Cell cycle analysis The H460 cells were serum starved without FBS overnight to synchronize cell phase, then the cells were treated with various concentrations of AZ4 for 24 h. The cells were harvested and incubated with hypotonic staining buffer [0.1% sodium citrate, 0.3% triton X-100, 0.01% propidium iodide (PI), and 0.01% ribonuclease A] for 15 min on ice in the dark. The DNA content was measured by a Becton Dickinson FACScan using Cell Quest software and analyzed using EXPO 32 and MultiCycle software (Beckman Coulter, High Wycombe, UK).
Microscopy image of caspase 3, 8, and 9 activity in H460 cells The method was according the report of Komoriya et al[15] with minor modifications. Briefly, the H460 cells were incubated with fluorogenic caspase substrates (PhiPhhiLux-G1D2, CaspaLux 8-L1D2, and CaspaLux 9-M1D2, OncoIm-munin Inc, Gaithersburg, MD, USA), GDEVDGI, IETDGI, and LEHDGI at 10 µmol/L respectively, while suspended in RPMI- 1640 plus 10% FBS, 10 mmol/L HEPES, and various concentrations of AZ4 for 24 h, 30 h(for caspases 8 and 9 only), and 48 h incubation. Substrates were present at 10 µmol/L throughout the course of the induction and imaging. The cells were viewed on a Nikon fluorescence microscope system (Tokyo, Japan). Samples were excited using a 488/518 nm krypton/argon laser, and fluorescent images were acquired. The brightness/contrast settings were adjusted so that the fluorescent signal of cells without AZ4 was near the background. As the fluorogenic caspase substrates are cleaved in apoptotic cells, cellular fluorescence shifts from below to above the fluorescence of the bulk solution in the same plane.
Western blot analysis of cyclin B, Cdc-2, p53, p21, and the Bcl-2 protein The method used was that of Bacus et al and slightly modified[16]. Briefly, the cells were collected from a 100 mm cultured dish after being challenged by various concentrations of the AZ4 compound at 24 and 48 h, respectively. The cell pellets were spun down by centrifugation at 1000×g for 20 min. The pellets were resuspended in cold buffer (10 mmol/L HEPES, pH 7.9), 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L diethiotreitol (DTT), 0.5 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L benzamidine, 30 mg/mL leupeptin, 5 mg/mL aprotinin, and 5 mg/mL pepstatin A, and incubated on ice for 5 min. The cells were broken down by lysis buffer and a sonicator. Cell lysates (25 µg) was separated by 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Amersham, Buckingshamshire, UK). The blots were incubated with blocking buffer (11 mmol/L Tris-vase pH 7.4, 154 mmol/L NaCl, and 5% skim milk), washed with washing buffer (11 mmol/L Tris-vase pH 7.4, 154 mmol/L NaCl, and 0.1% Tween-20), and incubated with specific antibodies against specific proteins cyclin B, Cdc-2, p53, p21, and Bcl-2. Each membrane was blocked in blocking buffer prior to incubation with antibodies. The primary antibody against cyclin B, Cdc-2, p53, p21, and Bcl-2, all belong to the mouse antihuman monoclonal antibody (Imgenex Co, San Diego, CA, USA). The secondary antibody (Jackson Immuno Research Lab Inc, West Grove, PA, USA) conjugated with horseradish peroxidase was added at an appropriate dilution by blocking buffer. The dilution factors for the primary antibodies were 1:500–1:1000 depending on the amount of proteins, and the dilution factors for the secondary antibodies were 1:2000. The primary β-actin antibody used was a mouse monoclonal antibody with the dilution factor of 1:10 000 (Biogenesis, England, UK). Immunodetec-tion was carried out using the enhanced chemiluminescence (NEN, Boston, MA, USA) detection system. To quantify the amount of protein expression achieved, we measured the intensity of chemiluminescence of the second antibody using a densitometer (BioRad Gel Doc 2000 software) and analyzed using Gel Doc (Hercules, CA, USA). The values in the relative protein interested expression-quantifying table represent the relative amount of protein expression in respect to β-actin expression divided by its control.
Results
Cytotoxic activity evaluation The chemical structures of AZ1 to AZ4 are shown in Figure 1. The chemical structure of AZ1 to AZ4 was composed of 2 aziridinylnaphthoquinones bridging by ethylene glycol units. AZ1 had the shortest ethylene glycol units; in retrospect, AZ4 contained the 4 ethylene glycol units. The cytotoxic activities of AZ1 to AZ4 against NSCLC H460 cells were evaluated; the human fetal lung fibroblast cell line (MRC5) was used as a normal cell control, and their IC50 values are listed in Table 1. The bis-aziridinylnaphthoquinone AZ1 to AZ4 were effective cytotoxic agents against H460 cells with IC50 values ranging from 1.23 to 7.21 µmol/L. The normal lung fibroblast cells MRC-5 were less sensitive to AZ1 to AZ4 with IC50 values of 2.8 to 12.7 µmol/L, respectively. The correlation between the cytotoxic activities and the linker distance between 2 aziridinylnaphthoquinone moieties against H460 cells was directly in proportion, but not with the MRC-5 cells. Initial characterization indicated that AZ4 was the most effective compound at inhibiting the growth of H460 cells and was less effective with MRC5 cells.
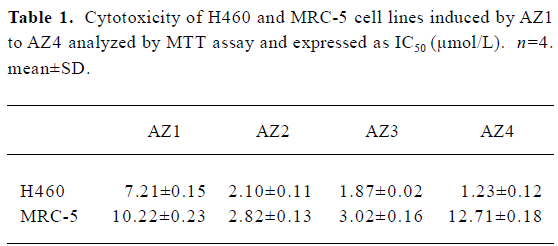
Full table
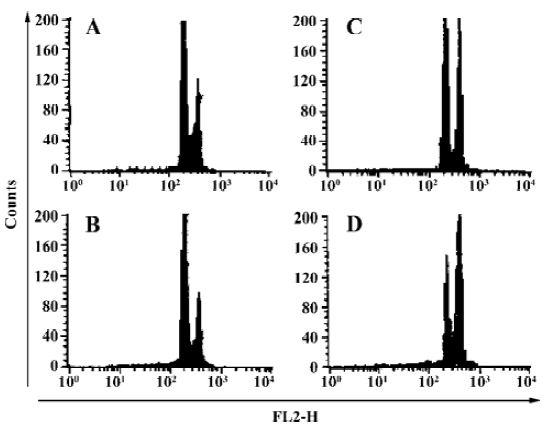
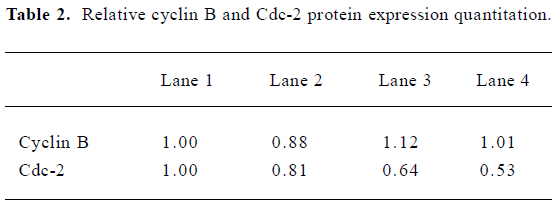
Induction of the G2-M cell cycle arrest and the effects on the expression of cell cycle-related proteins in H460 cells treated with AZ4 An analysis of DNA content was used to determine whether cell arrest was induced by AZ4. A DNA content analysis by a flow cytometer indicated that H460 cells had a significant population of cell arrest in G2-M treated with AZ4 in 24 h compared with 0.1% DMSO alone (Figure 2). The peak area under the G2-M phase started to ascend as low as 0.5 µmol/L with AZ4. The cell cycle-related proteins associated with mitotic, cyclin B and Cdc-2, in the AZ4-treated H460 cells for 24 h were also investigated (Figure 3, Table 2). The expression pattern of cyclin B in the cells remained basically unaltered with increased concentrations of AZ4 treatment; however, the expression pattern of Cdc-2 was decreased relative to the control in a dose-dependent manner. The expression of Cdc-2 decreased by 47% at 2.0 µmol/L with AZ4. We proposed that the growth arrest on H460 cells induced by AZ4 might be altered with reduced protein expression of Cdc-2, but not cyclin B.

Full table
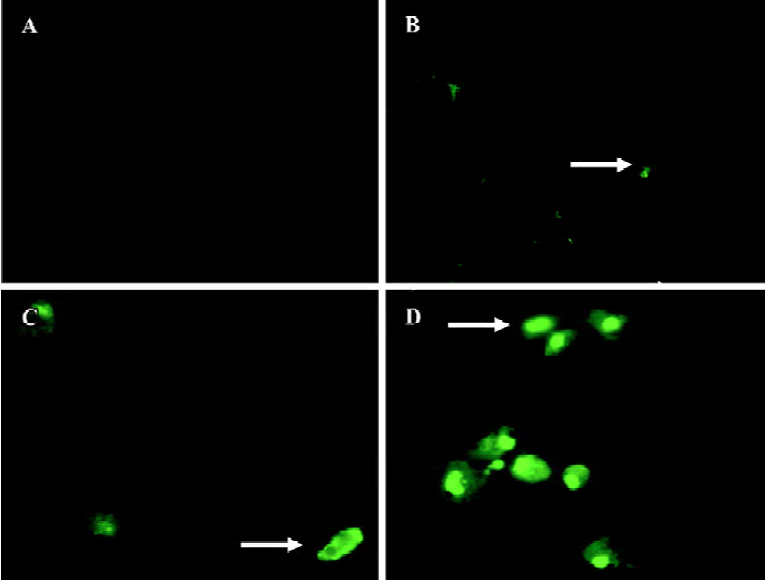
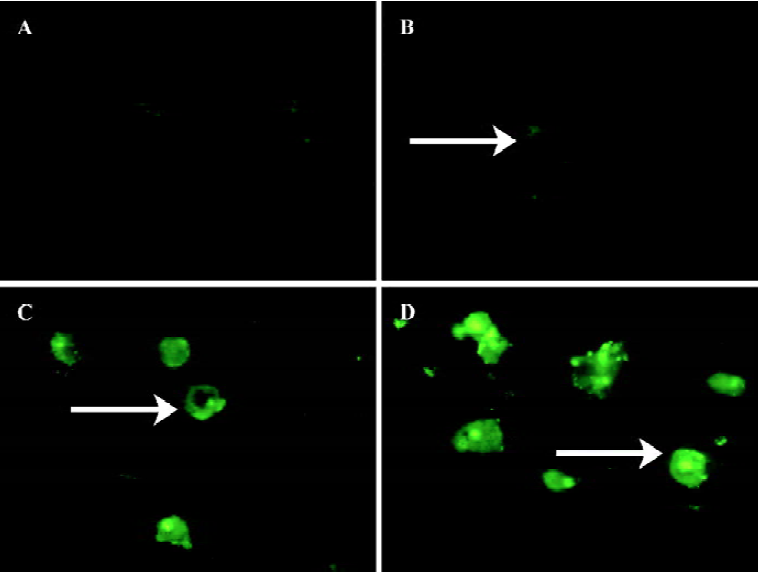
AZ4 triggers H460 cell apoptosis via caspase 3, 8, and 9-dependent routes and is associated with altered expression of p53 and Bcl-2 proteins We investigated whether apoptosis of H460 cells induced by AZ4 was mediated by caspase 3, 8, and 9 activation for 48 and 30 h after treatment. The activation of caspases 3, 8, and 9 was assessed by cell permeable fluorogenic caspase substrate, GDEVDGI, IETDGI, and LEHDGI, respectively. The induction of caspases 3, 8, and 9 would cleave the cell permeable to fluorogenic substrate individually, and then release the green fluorogenic substance. From those results, there was a large increase of the green peripheral clump as indicated by the arrows in Figures 4 and 5. The upstream enzyme of caspases 3 and 8 were activated in H460 cells induced by AZ4, but not incaspase 9 (data not shown). According to the visual observa-tion, the green peripheral clump appeared as low as the concentration at 0.625 µmol/L AZ4 at 30 h when observing caspase 8 (Figure 4) and 48 h for caspase 3 (Figure 5). However, enzyme activation of caspase 3 was not found to be as high as treated with the 2 µmol/L AZ4 (data not shown).
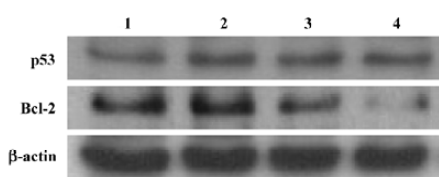
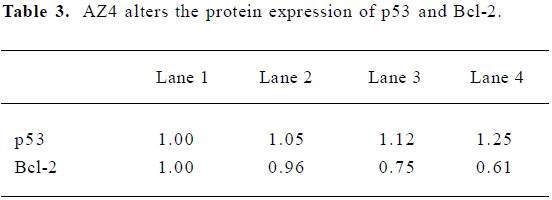
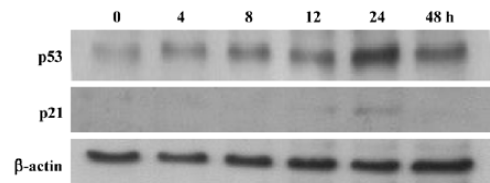
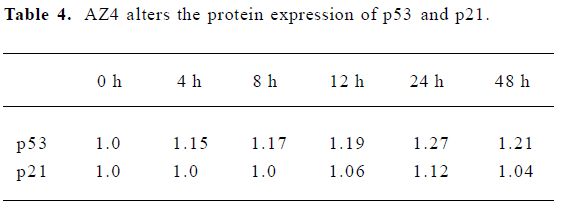
The apoptosis related proteins, tumor suppressor protein p53, and the anti-apoptotic protein Bcl-2 are two important components of the apoptotic pathway which act as regulators of apoptosis. From the results of the Western blot, AZ4 treatment altered both p53 and Bcl-2 expression in H460 cells in a dose-dependent manner after 48 h incubation (Figure 6, Table 3). The AZ4 induced overexpression of the p53 protein 25% more than the control when the H460 cells were exposed to 2.0 µmol/L AZ4. It is of interest that Bcl-2 protein expression was strongly reduced by 39% compared with the control when the H460 cells were treated with 2.0 µmol/L AZ4. In the time-dependent effect, the expression of p53 and p21 proteins were increased to a maximum at 24 h, and then decreased at 48 h (Figure 7, Table 4).
Full table
Full table
Discussion
Bioreductive drugs such as AZQ, mitomycin C, RH1 and TPZ, have been developed to exploit the oxygen deficiency in the hypoxic fraction of solid tumors on the premise that hypoxic cells should show a greater propensity for reductive metabolism than well-oxygenated cells[3,5–7]. Our pervious study on different series of bis-aziridinylnaphthoquinone compounds showed that these compounds exhibit more potent responses toward the solid tumors than the circulation tumors[14]. These results are supported by other reports indicating that there are differences in the reductive metabolism between the solid tumors and the circulation tumors[3]. Considering the importance of all the cellular reductases (eg NADPH cytochrome P450 reductase, cytochrome b5 reductase, NADP(H) oxidoreductase, NQO1) in response to the whole cellular reductive metabolism; these reductases are also responsible for the bioactivation of AZ4. The results from Table 1 suggest that AZ4 is a novel class of bis-aziridinylnaphthoquinone and is cytotoxic against H460 cells.
As shown in Figure 2, the cell cycle arrest at the G2-M phase was observed at 0.5 µmol/L AZ4. The G2-M phase-arresting proteins of H460 cells induced by AZ4 was related with the reduced expression of the Cdc-2 protein, but not cyclin B (Figure 3).
Apoptosis plays an important role in the removal of aberrant cells that might otherwise cause the development of tumors[17,18]. Normally, the process of cell reproduction is an ordered process known as cell cycle. The tumor suppressor gene p53 is a multifunctional protein mainly responsible for maintaining genomic integrity, and is the most frequently mutated gene in human tumors[19]. In response to DNA damage, aberrant growth signals or the chemotherapeutic drug p53 is stabilized and induces apoptosis and/or cell cycle arrest[20–22]. Therefore, p53 is a tumor suppressor gene with key regulator effects on both cell cycle and apoptosis. p53 also exerts its control on apoptosis by interacting with other important apoptotic molecules, such as members of the Bcl-2 family[23].
The protein from the Bcl-2 gene family, which is an anti-apoptotic associated protein, plays an important role in the regulation of apoptosis[24]. In the apoptotic pathway, some drugs will activate the cysteine protease family, such as caspases which specifically cleave their substrates after aspartic acid, then activate or inactivate their cellular protein targets by a process of limited proteolysis. The caspases play central roles in the execution of apoptosis[25–27]. Pro-apoptotic therapeutic strategies require, in turn, the exact knowledge of the very specific downstream pathway of apoptosis and of each single step in the targeted cell. In some cells, the signaling cascade is mediated by the caspases. Several proteins are known to regulate or interfere with this step of apoptosis, that is, proteins of the Bcl protein family, acting either anti-apoptotically (Bcl-2, Bcl-x etc) or pro-apoptotically (bax, bid etc)[28], and the pro-apoptotically-acting transcription factor p53.
The antitumor mechanism of AZ4 to lung cancer cell H460 was mediated with cell cycle arrest and the apoptosis pathway. According to the results shown in Figures 2 and 3, the cytotoxic activity of H460 cells induced by AZ4 was associated with G2-M phase arrest at 24 h. Then, the H460 cells steered into the apoptotic pathway after prolonged treatment with AZ4 for 48 h. We observed significant apoptosis phenomena in H460 cells associated with the activation of caspase 3 which mediated the caspase 8 enzymes, but not caspase 9, the upregula-tion of p53 protein, and Bcl-2 down regulation by various concentrations of AZ4 (Figures 4,5,6). Bcl-2 was inactivated and made the G2-M cells of H460 induced by AZ4 more susceptible to apoptosis which is supported by Yamamoto et al who proposed that stress response kinases phosphorylate Bcl-2 during cell cycle progression as a normal physiological process to inactivate Bcl-2 at the phase of G2-M[29]. To observe the timing relationship of protein expression from cell cycle arrest to apoptosis, the time-dependent effect (Figure 7), the expression of p53 and p21 proteins were increased to the maximum at 24 h, then decreased at 48 h. p21 is a member of the Cip/Kip family of proteins, which promote cell cycle arrest by binding to and inhibiting cyclin-dependent kinases. The latter, when coupled with specific cyclins, facilitates the orderly procession of the cell cycle. p21 is also tightly regulated at the transcriptional level by p53 and probably serves as the effector of the p53 cell cycle control[30]. This is in agreement with the above results, which support cell cycle arrest at 24 h, then apoptosis at 48 h.
In conclusion, AZ4 displayed selective cytotoxic activity against H460 cells and low cytotoxic activity when cultured with non-neoplastic human adherent lung fibroblasts. The cytotoxic effect induced by AZ4 on H460 cells may correlate with the induction of the G2-M cell cycle at 24 h which increased p53, p21, and Bcl-2 protein expression and decreased the expression of the Cdc-2 protein. The cells were then steered the apoptotic pathway for 48 h treatment, where the anti-apoptotic protein Bcl-2 and cell checkpoint proteins p53 and p21decreased, while the apoptosis enzymes caspase 3 and 8 were activated. It is believed that this cytotoxic mechanism study holds promise for the development of a new generation of potent antitumor agents.
References
- Pakunlu RI, Wang Y, Tsao W, Pozharov V, Cook TJ, Minko T. Enhancement of the efficacy of chemotherapy for lung cancer by simultaneous suppression of multidrug resistance and anti-apoptic cellular defense: novel multicomponent delivery system. Cancer Res 2004;64:6214-24.
- Brown JM. Tumor microenvironment and the response to anticancer therapy. Cancer Biol Ther 2002;1:453-8.
- Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumor hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev 2003;29:297-307.
- Naylor MA, Thomson P. Recent advances in bioreductive drug targeting. Mini-Rev Med Chem 2001;1:17-29.
- Phillips RM, Burger AM, Loadman PM, Jarrett CM, Swaine DJ, Fiebig HH. Predicting tumor responses to mitomycine C on the basis of DT-diaphorase activity or drug metabolism by tumorho-mogenares: implications for enzyme-directed bioreductive drug development. Cancer Res 2000;60:6384-90.
- Kim JY, Kim CH, Stratford IJ, Patterson AV, Hendry JH. The bioreductive agents RH1 and gamma-irradiation both cause G2/M cell cycle phase arrest and polyploidy in a p53-mutated human breast cancer cell line. Int J Rad Oncol Biol Phys 2004;58:376-58.
- Siim BG, Pruijn FB, Sturman JR, Hogg A, Hay MP, Brown JM, et al. Selective potentiation of hypoxic cytotoxicity of tirapaza-mine by its 1-N-oxide metabolite SR4317. Cancer Res 2004;62:736-42.
- Lin PS, Ho KC, Yang SJ. Tirapazamine (SR4233) interrupts cell cycle progression and induces apoptosis. Cancer Lett 1996;105:249-55.
- Lee AE, Wilson WR. Hypoxia-dependent retinal toxicity of bioreductive anticancer prodrugs in mice. Toxicol App Pharmacol 2000;163:50-9.
- Smitskamp-Wilms E, Giaccone G, Pinedo HM, Laan BFAM, Peters GJ. DT-diaphorase activity in normal and neoplastic human tissues; an indicator for sensitivity to bioreductive agents? Br J Cancer 1995;72:917-21.
- Danson S, Ward TH, Butler J, Ranson M. DT-diaphorase: a target for new anticancer drugs. Cancer Treat Rev 2004;30:437-49.
- Workman P, Walton MI. Enzyme directed bioreductive drug development. In: Fielden EM, Wardman P, editors. Selective activation of drug by redox processes. New York: Plenum Press; 1990. p173–91.
- Huang ST, Kuo HS, Lin CM, Tsai HD, Peng YC, Chen CT, et al. Synthesis and biological evaluation of novel bis-aziridinylna-phthoquinone derivatives. Oncology Res 2003;11:199-204.
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107-12.
- Komoriya A, Packard BZ, Brown MJ, Wu ML, Henkar PA. Assessment of caspase activities in intact apoptotic thymocytes using cell-permeable fluorogenic caspase substrates. J Exp Med 2000;191:1819-28.
- Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogen 2001;20:147-55.
- Dive C, Gregory CD, Phipps DJ, Evans DL, Miliner AE, Wyllic AL. Analysis and discrimination of necrosis and apoptosis (programmed cell death) by multiparameter flow cytometry. Biochim Biophys Acta 1992;1133:275-81.
- Scheer M, Kuebler AC, Zoller JE. Chemoprevention of oral squamous cell carcinomas. Onkologie 2004;27:187-93.
- Hsu S, Singh B, Schuster G. Induction of apoptosis in oral cancer cells: agents and mechanisms for potential therapy and prevention. Oral Oncol 2004;40:461-73.
- Koontongkaew S, Chareonkitkajorn L, Chanvitan A, Lee-lakriangsak M, Amornphimolttham P. Alteration of p53, pRb, cyclin D (1) and cdk4 in human oral and pharyngeal squamous cell carcinomas. Oral Oncol 2000;36:334-9.
- Schmitt CA, Low SW. Apoptosis and therapy. J Pathol 1999;197:127-37.
- Evans GL, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001;411:342-8.
- Pirollo KF, Bouker KB, Chang EH. Does p53 status influence tumor response to anticancer therapies? Anti-cancer Drugs 2000;11:419-32.
- Nylader K, Dabelsteen E, Hall PA. The p53 molecular and its prognostic role in squamous cell carcinomas of the head and neck. J Oral Pathol Med 2000;29:413-25.
- Kaul R, Mukherjee S, Ahmed F, Bhat MK, Chhipa R, Galande S, et al. Direct interaction with and activation of p53 by SMAR1 retards cell-cycle progression at G2/M phase and delays tumor growth in mice. Int J Cancer 2003;103:606-15.
- Gartel AL, Feliciano IL, Tyner A. A new method for determining the status of p53 in tumor cell lines of different origin. Oncol Res 2003;13:405-8.
- Nikitakis NG, Sauk JJ, Papanicolaou SI. The role of apoptosis in oral disease: Mechanisms; aberrations in neoplastic, autoimmune, infectious, hematologic, and developmental diseases; and thera peutic opportunities. Oral Surg Oral Med Oral Pathol Oral Radiol & Endodo 2004;97:476-90.
- Gibson LF, Fortney J, Magro G, Ericoson SG, Lynch JP, Landreth KS. Regulation of BAX and BCL-2 expression in breast cancer cells by chemotherapy. Breast Cancer Res Treat 1999;55:107-17.
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol Cell Biol 1999;19:8469-78.
- Karl A, Nath KA. Provenance of the protective property of p21. Am J Physiol Renal Physiol 2005;289:F512-3.