
Autophagy is involved in cytotoxic effects of crotoxin in human breast cancer cell line MCF-7 cells
Introduction
Autophagy is a dynamic process in which intracellular membrane structures sequester proteins and organelles for degradation in a lytic compartment. It is an evolutionarily, conserved process that occurs in all eukaryotic cells, from yeast to mammals[1,2]. Autophagy is mediated by double- or multi-membrane autophagosomes and can be induced under conditions of starvation or by certain hormones[3]. In addition to its basic role in the turnover of proteins and organelles, autophagy has multiple physiological and pathophysiological functions including cell differentiation, immune defense, and cell death[4]. Recent studies have found the activation of autophagy under pathological conditions, such as neurodegenerative diseases and hereditary myopathies[5,6]. While autophagy can represent an independent mechanism of cell self-destruction, named type II programmed cell death, it is often associated closely with apoptosis otherwise known as type I programmed cell death[7,8]. Autophagy and apoptosis can occur in the same cell concurrently or sequentially in response to the same stimulus. Autophagy exerts multiple impacts on apoptotic cell death, ranging from promoting to antagonizing apoptosis. In cells which have integrated caspase-dependent apoptotic signaling systems, autophagy can be triggered by mitochondrial membrane potential collapse and cytochrome c (cyto-c) redistribution following stimulation with apoptotic inducers[9–11]. However, the relationship between autophagic activation and caspase-independent apoptosis remains largely unknown. Moreover, studying the role of autophagy in cancer is at a very early stage[12], and even the most fundamental question –– whether autophagy kills cancer cells or protects them from unfavorable conditions –– has not been clearly answered.
Crotoxin (CrTX) is a cytotoxic PLA2 isolated from the venom of the South American rattlesnake, Crotalus durissus terrificus[13]. It is a non-covalent complex formed by 2 non-identical subunits, one acidic (subunit A) and one basic (subunit B). Subunit B is a PLA2 formed by a single chain of 122 amino acid residues cross-linked by 7 disulfide bonds[14]. Its cytotoxicity was independent of cell growth since both quiescent and proliferating cells had similar sensitivities. CrTX displays cytotoxic activity against a variety of murine[15] and human tumor cell lines in vitro[16] and appears to be highly active toward cell lines expressing a high density of epidermal growth factor receptors (EGFR)[17]; however, the precise mechanism of the cytotoxicity remains to be determined. It had previously been assumed that the toxin simply induces lysis through the disruption of the cell membrane.
Previous studies found that CrTX-induced death of human leukemic K562 cells was associated with the collapse of the mitochondrial membrane potential, the release of cyto-c, and the activation of caspase-3. Caspase inhibitors attenuated CrTX-induced K562 cell death. These studies provided evidence that an apoptotic mechanism contributes to the CrTX-induced death of K562 cells. CrTX also activated autophagy in these cells; however, the inhibition of autophagy potentiated the cytotoxicity of CrTX, suggesting autophagy delayed apoptosis in K562[18].
The human breast cancer cell line, MCF-7, which lacks procaspase-3 due to the functional deletion of the caspase-3 (CASP-3) gene, has been widely used in studying the importance of caspase-3 in apoptosis. Some studies found that caspase-3-mediated events appeared to be dispensable in determining the overall killing of cells, since 50 kb DNA fragments, chromatin condensation, Rb, gelsolin, and poly (ADP-ribose) polymerase (PARP) cleavage also occurred in caspase-3-deficient MCF-7 cells[19–21]. When caspase-3 has no contribution to cell death, cells can still die apoptotically by non caspase-3-dependent or caspase-independent mechanisms due to the activation of other effective caspases or the nuclear translocation of AIF[22].
To further investigate the relationship between molecular mechanisms of apoptosis and autophagy in tumor cells, this study focused on the role of autophagy in CrTX-induced cytotoxicity in MCF-7 cells where apoptosis is hampered due to the lack of caspase-3 activity.
Materials and methods
Drug preparation CrTX was supplied by Celtic Biotech Ltd (Dublin, Ireland). It was purified from Crotalus durissus terrificus venom by a combination of size exclusion and anion exchange. The identity of the protein was confirmed through molecular weight, determined by mass spectrometry (showing averaged signals at 9500 and 14500, in addition to the presence of isoforms). Purity was determined by PAGE and size exclusion was greater than 90%. Lethality in mice was determined by an ip injection of 0.1 mg with death being recorded within 3 h.
Cell culture Human breast cancer, MCF-7 cells were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were maintained in DMEM medium (Gibco, Rockville, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hangzhou Sijiqing Biological Engineering Material Co Ltd, Hangzhou, China), 0.03% l-glutamine (Sigma, St Louis, MO, USA), and 200 U/L insulin incubated in a 5% CO2 atmosphere at 37 °C and routinely subcultured every 2 or 3 d. Cells in a mid-log phase were used in the experiments. To determine the dose- and time-response of MCF-7cells to CrTX, the MCF-7 cells were plated into 96-well microplates (1.5×104 cells/well) and cultured for 14 h. CrTX (12.5, 25, 50, and 100 µg/mL) was added to the culture medium, and cell viability was assessed with an MTT assay 24, 48, and 72 h after drug treatment.
Determination of cell viability Cell viability was assessed by MTT assay[23] as described before. Briefly, when the MCF-7 cells confluenced by 70% in the 96-well microplates, different concentrations of CrTX were added in the absence or presence of the pan-caspase inhibitor, Z-Vad-fmk (Calbio-chem, La Jolla, CA, USA) ; 12.5, 25, and 50 μmol/L), for the indicated time. The MTT solution (5 mg/mL, Sigma) was added 4 h before the end of incubation and the reaction was stopped by 10% acidified SDS. The absorbance value (A) per well at 570 nm was read using an automatic multiwell spectrophotometer (Bio-Rad, Hercules, CA, USA). The ratio of cell death was calculated as follows: cell death (%)=(1-A of experiment well/A of control well)×100%.
Cytotoxicity assays Cytotoxicity was measured by LDH leakage using the LDH detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The samples of the supernatants and lysates were prepared as follows: when the MCF-7 cells confluenced by 70% in the 24-well micro-plates, the autophagy inhibitors 3-MA (10 mmol/L) or NH4Cl (10 mmol/L) were added 1 h before CrTX treatment. After being incubated for 24 h, the supernatants of the culture were collected following centrifugation. The cell pellets were lysed in 1% Triton X-100 at 37 °C for 45 min. Then the samples of supernatants and lysates were processed according to the instructions of the manufacturer. The absorbance value (A) at 400 nm was quantified by a spectrophotometer. LDH leakage was calculated as follows: LDH leakage (%)=(A positive–A positive blank)/(A negative-A negative blank)×100%.
Visualization of MDC-labeled vacuoles Autophagic vacuoles were labeled with monodansylcadaverin (MDC, Sigma)[24]. Briefly, the cells were seeded into microplates pre-exposed to 0.01% polylysine and incubated with CrTX (100 µg/mL) for 3, 6, and 12 h. Fresh DMEM containing 0.05 mmol/L MDC was added and the cells were incubated at 37 °C for another10 min. Then the cells were washed with phosphate buffered saline (PBS) 3 times and the microplates were inverted onto a slide and analyzed by fluorescence microscopy using an inverted microscope (Nikon Eclipse TE 300, Tokyo, Japan) equipped with a filter system (V-2A excitation filter: 380-420 nm, barrier filter: 450 nm). Microphotographs were taken with a Charge Coupled Device (CCD) camera.
Transmission electron microscope examination[25] After treatment with CrTX, the cells were fixed in ice-cold 2.5% glutaraldehyde in 0.1 mol/L PBS and preserved at 4 °C for further processing. When processing resumed, the cells were post fixed in 1% osmium tetroxide in the same buffer, dehydrated in graded alcohols, embedded in Epon 812, sectioned with ultramicrotome, stained with uranyl acetate and lead citrate, followed by an examination with a transmission electron microscope (Philips Tecnai 10, Eindhoven, the Netherlands).
Subcellular fractionation The isolation of mitochondria was performed as described by Qin et al[26]. Cells treated with CrTX for different times were harvested and rinsed with ice-cold PBS twice. The cells were suspended in buffer A (250 mmol/L sucrose, 1 mmol/L EDTA, 50 mmol/L Tris-HCl, 1 mmol/L DL-Dithiothreitol (DTT), 1 mmol/L phenylmethyl sulfonylfluoride (PMSF), 1 mmol/L benzamidine, 0.28 u/mL aprotinin, 50 µg/mL leupeptin, and 7 µg/mL pepstatin A, pH 7.4) and homogenated with a glass Pyrex microhomogenizer (30 strokes). The homogenates were centrifuged at 1000×g at 4 °C for 10 min, and the resultant supernatants were transferred to a new Eppendorf tube and centrifuged at 10000×g at 4 °C for 20 min to obtain the mitochondrial pellets and supernatants. The supernatants were transferred to a new tube and centrifuged at 100 000×g for 1 h at 4 °C to generate the cytosolic fraction. The mitochondrial pellet was washed 3 times in buffer B [250 mmol/L sucrose, 1 mmol/L ethylene-glycol bis(2-aminoethyl ether)tetraacetic acid (EGTA), 10 mmol/L Tris-HCl, 1 mmol/L DTT, 1 mmol/L PMSF, 1 mmol/L benzamidine, 0.28 u/mL aprotinin, 50 µg/mL leupeptin, and 7 µg/mL pepstatin A, pH 7.4] at 10 000 g for 10 min at 4 °C and then lysed in Western blot lysing buffer.
Protein preparation and immunoblotting The cells were harvested and rinsed with ice-cold PBS twice. Five volumes of Western blot lysing buffer for each volume of cell pellets was added and the mixture was sonicated on ice (1 s/mL per sonicate, resting wait for 30 s between intervals, 5 times), microcentrifugated at 10 000 rpm for 10 min, and the supernatants were preserved at -70 °C. Before immunoblotting, the protein content of each sample was adjusted by a bicin-choninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Proteins were separated on 12% SDS-PAGE gel, transferred to a nitrocellulose membrane and immunoblotted with goat polyclonal cathepsin B antibody (1:100; sc-6493, Santa Cruz, Santa Cruz, CA, USA), goat polyclonal cathepsin D antibody (1:100; sc-6486, Santa Cruz) or goat polyclonal cathepsin L antibody (1:100; sc-6499, Santa Cruz), and mouse monoclonal cyto-c antibody (1:1000; 7H8.2C12, PharMingen, San Diego, CA, USA) at 4 °C overnight. The primary antibodies were detected using horseradish peroxidase-conjugated anti-goat or anti-mouse antibody (Sigma) at a ratio of 1:5000 in blocking solution for 1 h at room temperature. Immunoreactivity was detected by enhanced chemiluminescence (ECL kit, Amersham Pharmacia Biotech, Piscataway, NJ, USA) and visualized by autoradiography. Protein β-actin (1:5000, A441, Sigma) and heat shock protein (HSP) 60 (1:2000, H3524, Sigma) were used as loading controls.
Immunofluorescence[27] The MCF-7 cells were seeded onto microplates and treated with CrTX (100 µg/mL). After fixation (methanol) and blocking (1% bovine serum albumin (BSA) dissolved in 0.1% Triton X-100), the cells were incubated with mouse polyclonal AIF antibody (1:100; PM-0215, Beijing Zhongshan Co Ltd, Beijing, China) at 4 °C overnight. A fluorescent fluorescein isothiocyanate (FITC) antibody (Sigma) was used to visualize the binding sites of the primary antibody. The cells were examined with a laser confocal microscope (Leica DMIRE2, Wetzlar, Germany).
Statistical analysis All data were presented as mean± SEM. Statistical analysis was carried out by ANOVA followed by Dunnett’s t-test, with P<0.05 considered signi-ficant.
Results
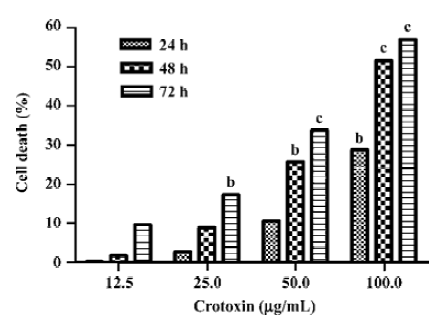
Inhibition of cell viability of MCF-7 cells by CrTX The cells were exposed to various concentrations of CrTX and various lengths of time. The cytotoxicity of CrTX was determined using the MTT assay. CrTX inhibited MCF-7 cell viability in a time- and dose-dependent manner. The MTT assay revealed that following 24 h of treatment, the inhibitory rate of CrTX (100 µg/mL) on MCF-7 cells was 28.85%± 0.01%, and when the incubation time was prolonged to 48 h, the inhibitory rate increased to 51.61%±0.03%, and then it reached 57.04%±0.04% after 72 h of treatment. At the dose of 50 μg/mL crotoxin, the inhibitory ratio was only 33.82%±0.04% with 72 h of treatment (Figure 1). Based on its effectiveness in inhibiting cell viability of MCF-7 cells, the CrTX concentration of 100 µg/mL was chosen for the subsequent experi-ments.
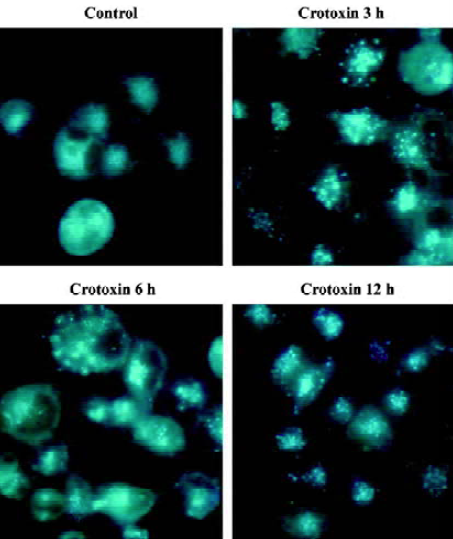
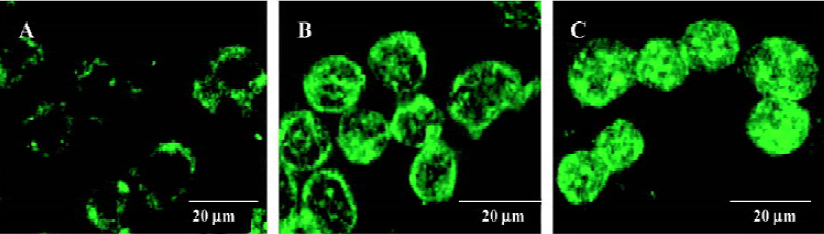
Vesicular accumulation of MDC after CrTX treatment The autofluorescent substance MDC has recently been shown to be a specific marker for autophagic vacuoles (AVs). When the cells were viewed with a fluorescence microscope, MDC-labeled AVs appeared as distinct dot-like structures distributing in the cytoplasm or in the perinuclear regions. It was found that there was no notable difference in the number of vesicles after 0.5 h of CrTX treatment (data not shown). When the duration of CrTX treatment was extended to 3 h, an increase in the MDC-labeled vesicles was observed as indicated by the appearance of punctuate MDC fluorescence, suggesting an induction of AVs formation after CrTX treatment. Increased vacuolar accumulation of MDC continued during the 12 h of the CrTX treatment (Figure 2).
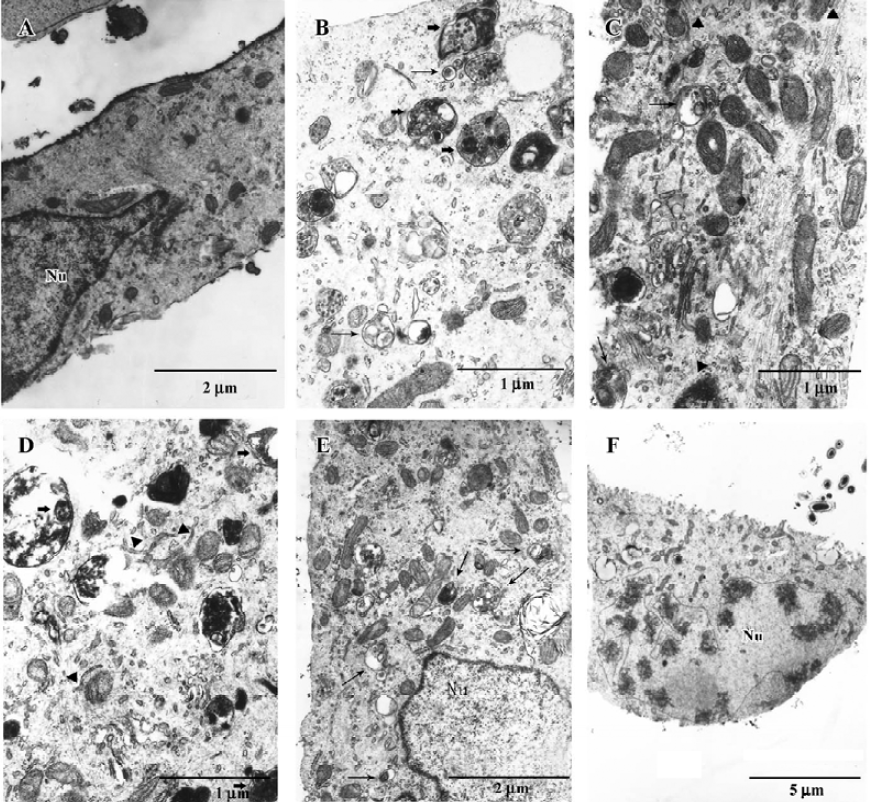
Ultrastructural examination of autophagy after CrTX treatment MCF-7 cells are very useful in the study of apoptotic-autophagic relationships because they may enter both the apoptotic and the autophagic pathways of cell death. The ultrastructural changes in MCF-7 cells during CrTX treatment were evaluated with the use of a transmission electron microscope (TEM). The untreated cells exhibited normal ultrastructural morphology of cytoplasm, cell organelles, and nuclei (Figure 3A). CrTX treatment for 6 h resulted in the formation of many autophagosomes and the sequestration of cytoplasm portions and organelles by double membranes, possibly derived from the endoplasmic reticulum. Double-membraned, giant autophagosomes filled with organelles were observed. Mitochondrial swelling and autolysosomes were also frequently observed. These processes preceded apoptotic morphology including nuclear envelope umbilication and chromatin compaction and pyknosis, features of caspase-independent apoptosis (Figure 3B–3F).
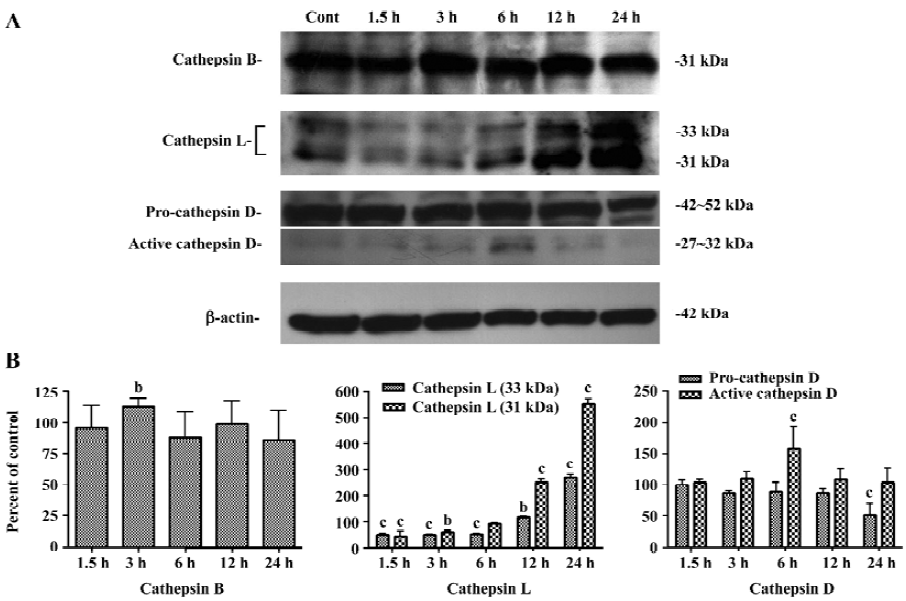
Increases in protein levels of cathepsin B, D, and L after CrTX treatment Since the lysosomal proteases are involved in both apoptosis and autophagy, they (or their targets) may serve as molecular links between both programmed cell death (PCD) types in tumor cells. Western blotting results revealed that a transient increase in cathepsin B levels was observed within 3 h following CrTX treatment. Cathepsin L decreased initially following CrTX treatment, but markedly increased 12 h later; pro-cathepsin D showed little change after CrTX treatment, but decreased dramatically 24 h later. Processed cathepsin D (active cathepsin D) was maintained at a high level 6 h after CrTX treatment (Figure 4).
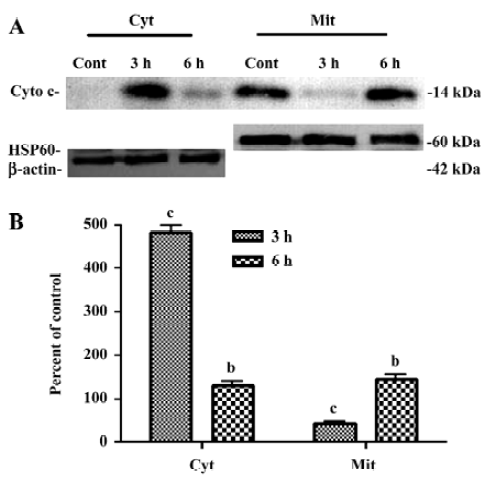
Redistribution of cyto-c and AIF with CrTX treatment As mitochondrial swelling induced by CrTX was observed by TEM, immunoblotting and immunofluorescence were used to investigate the localization of cyto-c and AIF after CrTX treatment. Cyto-c was released from the mitochondria into the cytoplasm 3 h after CrTX treatment (Figure 5). Confocal microscopy was employed to evaluate the changes of AIF cellular localization. AIF was released from the mitochondria and transferred into the nucleus as early as 1.5 h after CrTX treatment; AIF aggregated in the nucleus and displayed a tendency for uneven distribution (Figure 6).
Effects of autophagy and caspase inhibitors on the cytotoxicity of CrTX It has been reported that LDH leakage not only occurs during necrosis, but also during the process of apoptosis[28,29]. Since 3-MA interferes with the MTT assay, LDH leakage was measured as an index of cell death after co-treatment with CrTX and the autophagic inhibitors 3-MA (10 mmol/L) and NH4Cl (10 mmol/L). 3-MA, or NH4Cl individually had no significant effect on LDH leakage; however, pretreatment of MCF-7 cells with 3-MA and NH4Cl significantly decreased the LDH leakage induced by CrTX (Figure 7A).
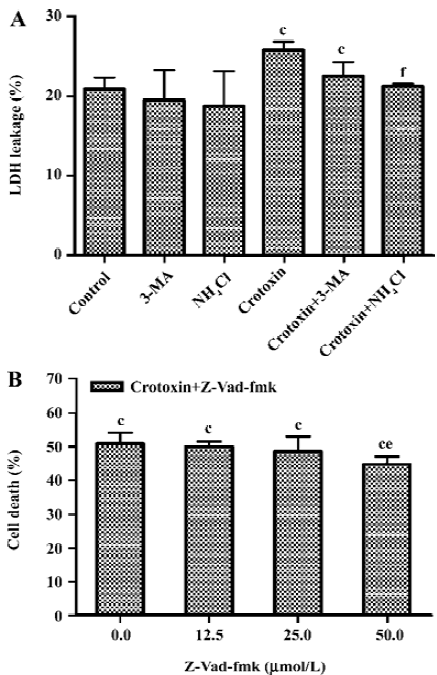
As caspases other than caspase-3 may contribute to apoptotic death in MCF-7 cells after CrTX treatment, the effects of pretreatment with the pan-caspase inhibitor, Z-Vad-fmk, were assessed. As shown in Figure 7B, Z-Vad-fmk slightly, but significantly inhibited the cytotoxicity of CrTX.
Discussion
CrTX is a potent antitumor agent and displays cytotoxic activity against a variety of murine and human tumor cell lines in vitro[15,16]. It requires both the PLA2 activity of subunit B[15] and the ability of the complex to dissociate into their subunits[15,29]. CrTX-induced cytotoxic effects appear to be highly active in cell lines expressing a high density of EGFR[17], thus suggesting that EGFR, or a receptor function, play a role in targeting. Antitumor efficacy in vivo, using a daily im administration of CrTX, has been demonstrated on Lewis lung carcinoma (83% growth inhibition) and MX-1 human mammary carcinoma (69% growth inhibition). A lower activity (44% growth inhibition) was observed with HL-60 leukemia cells, suggesting that CrTX may have a certain specificity toward solid tumors[30].
Our previous studies have revealed that the mechanism of the cytocidal action of CrTX may be more complex than what has been previously thought. It has been demonstrated that both autophagy and apoptosis were activated during the CrTX-induced death of K562 leukemia cells, but autophagy seemed to delay the onset of apoptosis[18].
Autophagy is biochemically and morphologically distinct from apoptosis, and is controlled by a different set of genes. Malignant transformation is frequently associated with the suppression of autophagy[31]. It has also been revealed that the silencing of the pro-apoptotic gene BID in MCF-7 cells led to the inhibition of apoptosis and a shift of cell death towards autophagy[32]. The ability to induce autophagy in cancer cells could be especially important in apoptosis-resistant cell lines, where pro-apoptotic genes were mutated. It is postulated that autophagic cell death induced by some anticancer agents underlines their potential as a new cancer therapy modality[31]. The present observations using electron microscopy revealed many isolated membranes with double or multi-membrane autophagosomes engulfing cytoplasma fractions and organelles 3 h after CrTX treatment. Simultaneously, there was a notable increase in MDC-stained vesicles following 3 h of CrTX treatment, which continued even after 12 h of CrTX exposure. The blockage of autophagy by the autophagic inhibitor, 3-MA, partially inhibited CrTX-induced cell death. These studies suggest that autophagy plays an important role in the CrTX-induced death of MCF-7 cells.
Increasing evidence suggests that lysosomes are important mediators of PCD. In autophagic cell death, lysosomes fuse with autophagosomes to form autophagolysosomes in which the cell contents are degraded[33]. Cathepsins trigger apoptosis via a number of pathways including directly activating caspases, and cleave BID[34,35]. Recently, studies also showed that if cathepsins were inhibited by a cathepsins inhibitor E64d in MCF-7 cells treated with camptothecin (CPT), CPT-induced BAX and BID aggregation on mitochondria were prevented and apoptosis was significantly reduced. This was accompanied by an increase in autophagosome formation[32]. The present observation, using Western blot analysis, showed a progressive increase in cathepsin B activity 3 h after CrTX treatment, whereas cathepsin L decreased initially, but markedly increased 12 h following CrTX treatment. Pro-cathepsin D showed no change with CrTX treatment, but processed cathepsin D (active cathepsin D) was maintained at a very high level 6 h after CrTX treatment. The activation of the lysosomal enzyme could be blocked by NH4Cl. The effects of NH4Cl further added evidence supporting an involvement of autophagy in the antitumor action of CrTX.
Recently, several lines of evidence demonstrated that the mitochondrion was the centre in cross-talk between autophagy and caspase-independent apoptosis[36]. The present results demonstrated that cyto-c release occurred as early as 3 h after CrTX treatment. However, when the treatment time prolonged to 6 h, the release of cyto-c decreased suggesting that the effects of CrTX on mitochondria were transient. MCF-7 cells are deficient in caspase-3 activity. Since caspase-3 is a major executioner caspase and plays very important roles in apoptosis, it is speculated that caspase-dependent apoptosis in MCF-7 cells would be seriously hampered. Some studies showed that MCF-7 cells underwent caspase-independent cell death. Autophagy thus plays a role in MCF-7 death[32,37]. The present study found that AIF was relocated from the mitochondrion into the nuclei 1.5 h after CrTX treatment. AIF is a phylogenetically conserved, mitochondrial intermembrane flavoprotein that has the ability to induce apoptosis via a caspase-independent pathway. AIF in the cytoplasm positively feedbacks to mitochondria, leading to a drop in mitochondrial membrane potential and the release of cyto-c[38,39]. In the present study, the features of caspase-independent apoptosis including nuclear envelope umbilication and chromatin compaction were also observed after 6 h of CrTX treatment. Pretreatment of MCF-7 cells with the pan-caspase inhibitor, Z-Vad-fmk, slightly but significantly inhibited the cytotoxicity of CrTX, suggesting that an apoptotic mechanism also contributed to cell death induced by CrTX.
In summary, both autophagy and caspase-independent apoptosis participated in cell death in the caspase-3-deficient MCF-7 cells following treatment with CrTX. Autophagy (PCD type II) may be the dominant mechanism of MCF-7 cell death induced by CrTX. The precise mechanisms underlying the different roles of autophagy in cell death remain to be studied. It is important to investigate the interactions between the autophagy and apoptotic signal pathways. Such studies will help us understand the role of autophagy in cell death and survival and provide new insights into antitumor chemotherapy.
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004;6:463-77.
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 2004;36:2445-62.
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun 2004;313:453-8.
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000;290:1717-21.
- Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol 2002;17:897-908.
- Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci 2003;3:64-9.
- Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol 2004;36:2405-19.
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 2004;16:663-9.
- Bursh W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 2001;8:569-81.
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366:177-96.
- Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res 2001;268:139-49.
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005;5:726-34.
- Faure G, Harvey AL, Thommson E, Saliou B, Radvanyi F, Bon C. Comparison of crotoxin isoforms reveals that stability of the complex plays a major role in its pharmacological action. Eur J Biochem 1993;214:491-6.
- Aird SD, Kaiser II, Lewis RV, Kruggel WG. A complete amino acid sequence for the basic subunit of crotoxin. Arch Biochem Biophys 1986;249:296-300.
- Corin RE, Viskatis LJ, Vidal JC, Etcheverry MA. Cytotoxicity of crotoxin on murine erythroleukemia cells in vitro. Inves New Drugs 1993;11:11-5.
- Rudd CL, Viskatis LJ, Vidal JC, Etcheverry MA. In vitro comparison of cytotoxic effects of crotoxin against three human tumors and a normal human epidermal keratinocyte cell line. Invest New Drugs 1994;12:183-4.
- Donato NJ, Martin CA, Perez M, Newman RA, Vidal JC, Etcheverry MA. Regulation of epidermal growth factor receptor activity by crotoxin, a snake venom phospholipase A2 toxin. Biochem Pharmacol 1996;51:1535-43.
- Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P, Qin ZH. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006;47:521-30.
- Xue LY, Chiu SM, Oleinick NL. Photodynamic therapy-induced death of MCF-7 human breast cancer cells: a role for caspase-3 in the late steps of apoptosis but not for the critical lethal event. Exp Cell Res 2001;263:145-55.
- Mathiasen IS, Lademann U, Jaattela M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res 1999;59:4848-56.
- Janicke RU, Ng P, Sprengart ML, Porter AG. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem 1998;273:15540-5.
- Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, et al. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J 2006;25:4061-73.
- Mosman T. Rapid colorimetric assay for cellular growth and survival: application and cytotoxicity assays. Immunol Methods 1983;65:55-63.
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 1995;66:3-14.
- Fengsrud M, Roos N, Berg T, Liou W, Slot JW, Seglen PO. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Exp Cell Res 1995;221:504-19.
- Qin ZH, Wang YM, Kikly KK, Sapp E, Kegel KB, Aronin N, et al. Pro-caspase-8 is predominantly localized in mitochondia and released into cytoplasm upon apoptotic stimulation. J Biol Chem 2001;276:8079-86.
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 2002;158:507-17.
- Thomas CE, Mayle DA. NMDA-sensitive neurons profoundly influence delayed staurosporine-induced apoptosis in rat mixed cortical neuronal cultures. Brain Res 2000;884:163-73.
- Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci 1997;9:159-69.
- Newman RA, Vidal JC, Viskatis LJ, Johnson JI, Etcheverry MA. VRCTC–310 –– a novel compound of purified animal toxins separates antitumor efficacy from neurotoxicity. Invest New Drugs 1993;11:151-9.
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004;23:2891-906.
- Lamparska-Przybysz M, Gajkowska B, Motyl T. Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF-7 cells exposed to camptothecin. J Physiol Pharmacol 2005;56 Suppl 3:159-79.
- Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Ann Rev Biochem 2000;69:303-42.
- Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM, et al. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 2004;279:3578-87.
- Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem 2001;276:3149-57.
- Ferri KF, Jacotot E, Blanco J, Este JA, Zamzami N, Susin SA, et al. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: role of mitochondria and caspases. J Exp Med 2000;192:1081-92.
- Gorka M, Daniewski WM, Gajkowska B, Lusakowska E, Godlewski MM, Motyl T. Autophagy is the dominant type of programmed cell death in breast cancer MCF-7 cells exposed to AGS 115 and EFDAC, new sesquiterpene analogs of paclitaxel. Anticancer Drugs 2005;16:777-88.
- Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): Key to the conserved caspase-independent pathways of cell death? J Cell Sci 2002;115:4727-34.
- Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, et al. Dominant cell death induction by extramito-chondrially targeted apoptosis-inducing factor. FASEB J 2001;15:758-67.