
Proteomic analysis of HuH-7 cells harboring in vitro-transcribed full-length hepatitis C virus 1b RNA1
Introduction
Hepatitis C virus (HCV) is a major worldwide health problem. In total, 70%–90% of people who become infected fail to have the virus removed and remain chronically infected with the risk of developing liver cirrhosis and hepatocellular carcinoma[1]. Thus far there is no prophylactic vaccine to prevent HCV infection, and the current treatment, α-interferon in combination with ribavirin, is not satisfactory because of significant side-effects and resistance.
The genome of HCV is a single-stranded, positive-sense RNA molecule approximately 9600 nucleotides in length. It consists of a 5´ untranslated region (UTR), a single large open reading frame, and a 3’ UTR[2]. The 5’ UTR is required for translation of positive-strand viral RNA and replication of negative-strand RNA. The single open reading frame encodes a large viral polyprotein of ~3000 amino acids[3] which is cleaved co- and post-translationally into at least 10 individual polypeptides with the following gene order: 5’-C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B-3’[4]. The 3’ UTR is closely related to virus replication and assembly[5] and is comprised of 3 domains: a variable, type-specific region followed by a (U) C-rich sequence of variable length, and a highly-conserved 98 nucleotide sequence designated the X tail at the 3’ end[6]. In vivo studies have demonstrated that the X tail is essential for replication[7].
Because of the lack of efficient and accurate in vitro models, basic studies defining the role of regulatory elements for replication and testing novel anti-HCV substances have been significantly hampered. Although infection of primary cells or cell lines with high-titer HCV-containing sera is possible, replication levels in these cultures are too low to allow detailed studies of HCV replication[8]. As an alternative, the development of selectable subgenomic HCV replicons has enabled the study of viral RNA replication in cell culture[9]. The selection of these RNA, which lack the complete structural region from gene C up to either p7 or NS2, was made possible by the insertion of the gene encoding neomycin phosphotransferase (neo) downstream of the HCV IRES, with translation of the HCV NS proteins directed by the IRES of Encephalomyocarditis virus. Although replication levels of these RNA were high, subgenomic HCV replicons were insufficient to study what happens in HCV-infected cells due to the lack of the HCV structural proteins and potential effects on cellular metabolism that could be associated with their presence. A genome-length HCV-RNA replication system, however, would allow in-depth study of the alterations that HCV-infected cells undergo. Several genome-length HCV-RNA replication systems, using the N, Con-1, O, and H77 strains, have been reported[10–12]. However, at present, the 3’ end of the HCV genome sequence in GenBank generally terminates at the polyadenylation site. Therefore, previous work has not taken into account the final 3’ 98 conserved nucleotides in the construction of cDNA of the HCV genome. This may be one of the main causes as to why whole HCV replicon cannot be efficiently replicated in such experiments.
In order to ensure that HCV-RNA was able to replicate, transcribe, and synthesize proteins effectively in cell culture, the complete genome of HCV was used, including the 98 nucleotides at the 3' end in the present study. Our study is to discuss the proteomic techniques have been applied to globally analyze the protein expression profiles of a human hepatocarcinoma cell line HuH-7 harboring in vitro-transcribed HCV 1b full length RNA (HuH-7–HCV) including the X tail. This work was aimed at elucidating the components of HCV replication and the cellular responses to HCV replication.
Materials and methods
Cells HuH-7 cells were grown in the presence of 5% CO2 in Dulbecco’s modified Eagle’s medium with high levels of glucose (0.45%) supplemented with 10% fetal calf serum and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin).
RNA preparation and transfection A recombinant expression plasmid, pSP72-HCV, containing the full-length HCV 1b genome, including the 98 nucleotides at the 3' end was constructed as described[13]. The plasmid DNA was extracted by phenol/chloroform extraction and ethanol precipitation. The HCV genomic RNA was transcribed by using MEGAscript TM T7 kit (Ambion, USA) in accordance with the manufacturer’s protocol. For a 20 µL reaction mixture, transcription was stopped by the addition of 4 U of DNase I and the mixture was incubated for 45 min at 37 ℃. RNA was extracted with 1 mL TRIzol reagent (Invitrogen, USA) and precipitated with 500 µL isopropanol. RNA pellets were washed once with 75% ethanol and dissolved in RNase-free water. To remove the residual amount of template DNA, RNA preparations were extracted once with acid phenol, precipitated with ethanol, and resuspended in RNase-free water.
HuH-7 cells were transfected with the RNA by electroporation and designated as HuH-7–HCV. Briefly, 5 µg RNA was mixed with 2×106 cells suspended in 500 µL phosphate-buffered saline (PBS) in a cuvette with a gap width of 0.2 cm (Bio-Rad, Hercules, CA, USA). Electroporation consisted of 2 pulses of currents delivered by the Gene pulser II electroporation device (Bio-Rad, USA), which was set at 1.5 kV, 25 µF and maximum resistance. Mock-transfected (electroporation without HCV genomic RNA) cells were used as a control and designated as HuH-7 mock. The culture media of both HuH-7–HCV and HuH-7 mock cells were removed completely after 24 h and stored at 4 ℃ for subsequent assays. The cells were washed twice with PBS prior to refeeding with fresh culture media. The culture media of HuH-7–HCV and HuH-7 mock cells were collected at 48, 72, and 96 h post-transfection respectively. At each time point, 3 samples from different plates were collected.
Detection of HCV accumulation in culture media by FQ–PCR and Western blotting HCV-RNA titers in the culture media were analyzed by a HCV fluorescence PCR diagnostic kit (DaAn Gene, China).
HCV core and the NS3 protein were detected by Western blotting. The 96-h post-transfection culture media of both HuH-7–HCV and HuH-7 mock cells were separated by 10% SDS–PAGE, electrotransferred to nitrocellulose membranes (Millipore, USA), and blocked with 5% non-fat milk in TBST. The membranes were immunoblotted with rabbit anti-HCV core (1:200) and an anti-HCV–NS3 polyclonal antibody (1:200; BIOS, China). Secondary antirabbit antibodies conjugated to horseradish peroxidase (BIOS, China) were applied at a dilution of 1:2000. The signals were detected by an ECL detection reagent (Pierce, USA).
2-D electrophoresis At 96 h post-transfection, the HuH-7–HCV and HuH-7 mock cells were harvested and dissolved in lysis buffer [7 mol/L urea, 2 mol/L thiourea, 1% Triton X-100, 4% (w/v) CHAPS, 4% Tris-base, 40 mmol/L dithiothreitol, 2% pharmalyte, 1 mmol/L phenylmethyl-sulfonyl fluoride, 1 g/L DNase I and 0.25 g/L RNase A]. The lysates were vortexed and incubated at 37 ℃ for 1 h and centrifuged at 1000×g for 30 min at 4 ℃. First-dimension isoelectric focusing was carried out using an Ettan IPGphor isoelectric focusing system, as described by the manufacturer (GE Amersham). Samples containing up to 100 µg protein were added to 13 cm IPG strips (pH 3−10). The strips were then applied by rehydration at 30 V for 12 h, and 1 h at 500 V. A gradient was then applied at 1000 V for 1 h, and 8000 V for 8 h. The temperature throughout this process was maintained at 20 ℃. Separation in the second dimension was carried out at a current setting of 15 mA/gel for the initial 20 min and 30 mA/gel until the bromophenol blue front reached the bottom of the gels. To account for experimental variation, 3 batches of total proteins extracted from cells were subjected to 2-D electrophoresis (2DE) and replicate gels were simultaneously run 3 times. The gels were stained, respectively, with Coomassie brilliant blue and silver nitrate, as described previously[14]. Protein patterns in the gels were recorded as digitalized images using UMax powerlook 2110XL (GE Amersham, USA) scanner.
In-gel protein digestion In-gel digestion of proteins was carried out as follows. The spots of interest were excised and put into 1.5-mL microtubes. Samples were destained with 200−400 µL of 100 mmol/L NH4HCO3/30% ACN and dried in a vacuum centrifuge. The dried gel pieces were rehydrated in 5 µL of 10 ng/µL trypsin (TPCK-treated, proteomics grade, Promega, USA), and incubated at 4 ℃ for 30−60 min. After digestion, 30 µL of 25 mmol/L NH4HCO3 was added, and the tubes were incubated at 37 ℃ for 20 h. The supernatants were recovered, and the extractions were repeated with 100 µL of 60% ACN/0.1% TFA. The extracted solutions were mixed and dried in a vacuum centrifuge.
MALDI–TOF MS analysis The peptide mixtures were solubilized with 5 µL of 1% formic acid, then vortexed and centrifuged. Samples (2 µL) were spotted on the target and dried. In total, 0.5 µL saturated acyano-4-hydroxy-trans-cinnamic solution was spotted on the samples and air dried. Spots were then analyzed in a Bruker-Daltonics AutoFlex TOF–TOF LIFT mass spectrometer (Bruker, Germany). The parameters were set up as follows: positive ion-reflector mode, accelerating voltage 20 kV. A trypsin-fragment peak served as an internal standard for mass calibration. A list of the corrected mass peaks was obtained as PMF. Proteins were identified from PMF data using MASCOT software (http://www.matrixscience.com). The following search parameters were applied: protein mass unrestricted; peptide mass tolerance±100 ppm, fragment mass tolerance±0.8 Da, number of missed cleavage sites allowed 1, cysteine residue modified as carbamidomethyl-cys, variable modifications oxidation (M), mass values MH+, and monoisotopic. NCBI were used as the protein sequence databases.
Real-time RT–PCR and Western blotting analysis for differentially expressed proteins To validate the MS results, another set of cells was isolated for total mRNA and subsequent real-time RT–PCR. The cDNA was synthesized by random hexamer primed reverse transcription using the RevertAid first strand cDNA synthesis kit (Fermentas, Canada). Real-time PCR was performed using the ABI 7500 real-time PCR system and SYBR premix ex Taq (perfect real time; TaKaRa, Dalian, China), primarily following the manufacturers’ protocol. In brief, the reaction mixture (25 µL total volume) contained 500 ng cDNA, primers (Table 1) at 0.2 µmol/L final concentration and 12.5 µL SYBR premix ex Taq. Thermal cycling conditions were 95 ℃ for 10 s and 40 cycles at 95 ℃ for 5 s and 60 ℃ for 34 s. The experiments were performed in triplicate in the same reaction. Human GAPDH gene was amplified as an internal control in the same reaction with target genes. Comparative quantification was determined using the 2ΔCt method[15].

Full table
An equal quantity of protein was used for the Western blotting analysis of the glucose-regulated protein 78 kDa (GRP 78) and keratin 9 protein expressions. Cells were lysed in RIPA buffer with protease inhibitors. Western blot analysis was performed as described earlier, and β-actin was used as an internal control. Rat anti-GRP 78 and goat anticytokeratin 9 (Santa Cruz, Santa Cruz, CA, USA) were diluted at 1:500, while a rabbit anti-β-actin polyclonal antibody (BIOS, China) was diluted at 1:200. Secondary antirat, antigoat, and antirabbit antibodies conjugated to horseradish peroxidase (BIOS, China) were applied at 1:2000 dilution.
Statistical analysis The expression levels of the proteins were quantified by analyzing the intensity of each spot with PDQuest software (Bio-Rad, USA). The differences in the protein expression levels between HuH-7–HCV and HuH-7 mock cells were analyzed by Student’s t-test or Mann-Whitney U-test. Probability values of less than 0.05 were considered significant. All analyses were carried out using SPSS 10.0 statistical software (SPSS, Chicago, IL, USA).
Results
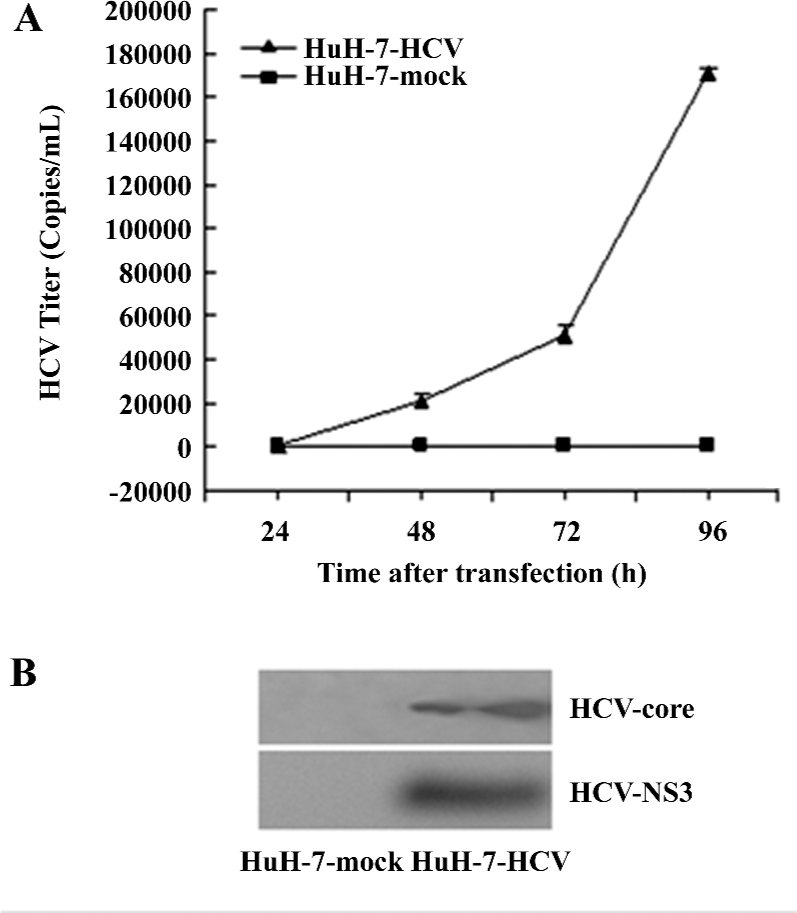
HCV-RNA and protein accumulation in HuH-7–HCV culture media Following transfection, increasing titers of HCV-RNA ranging from a few copies to 1×106 copies/mL were detected in the HuH-7–HCV cell culture media by fluorescence RT–PCR assays. These levels increased significantly over with time post-transfection. In contrast, in the HuH-7 mock culture supernatants, the HCV titers were <80 copies/mL at 24, 48, 72, and 96 h post-transfection, respectively (Figure 1A). Similarly, HCV core and the NS3 protein were detected in the culture media of HuH-7–HCV by Western blotting, but not in that of HuH-7 mock cells (Figure 1B). These results demonstrated that HCV-RNA was efficiently replicated in the in vitro culture system.
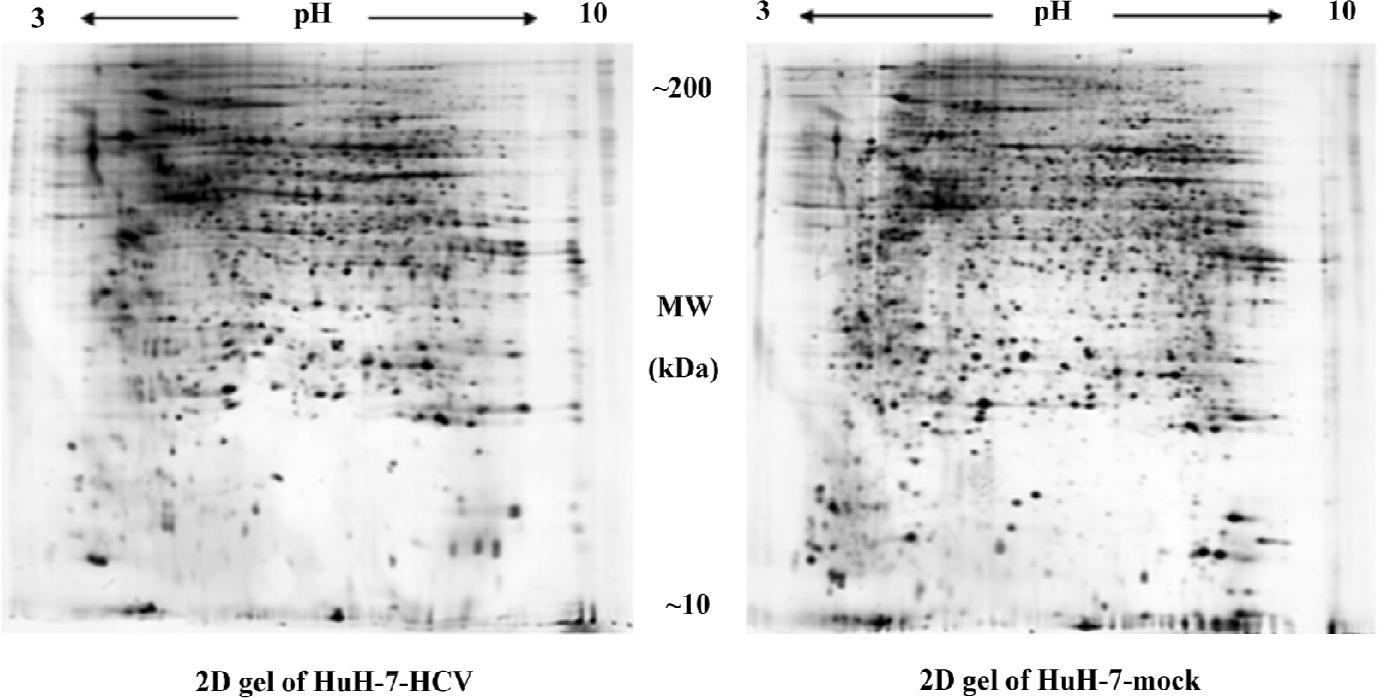
Proteomic analysis of differentially-expressed proteins The proteome of the HuH-7–HCV and HuH-7 mock cells was compared using 2DE (Figure 2). Reproducible high-resolution 2DE profiles were obtained. The average numbers of spots representing either individual or similarly migrating protein species obtained with both HuH-7–HCV and HuH-7 mock cells were 2618±111 and 2410±248, respectively. Fifty-four spots were found to be significantly altered (P<0.05). Among them, the intensities of 29 protein spots were increased and the intensities of 25 protein spots were decreased in HuH-7–HCV cell extracts as compared to those obtained with HuH-7 mock cell extracts. The 10 spots showing the greatest increase in signal and the 10 spots showing the greatest decrease in signal were chosen for further analysis.
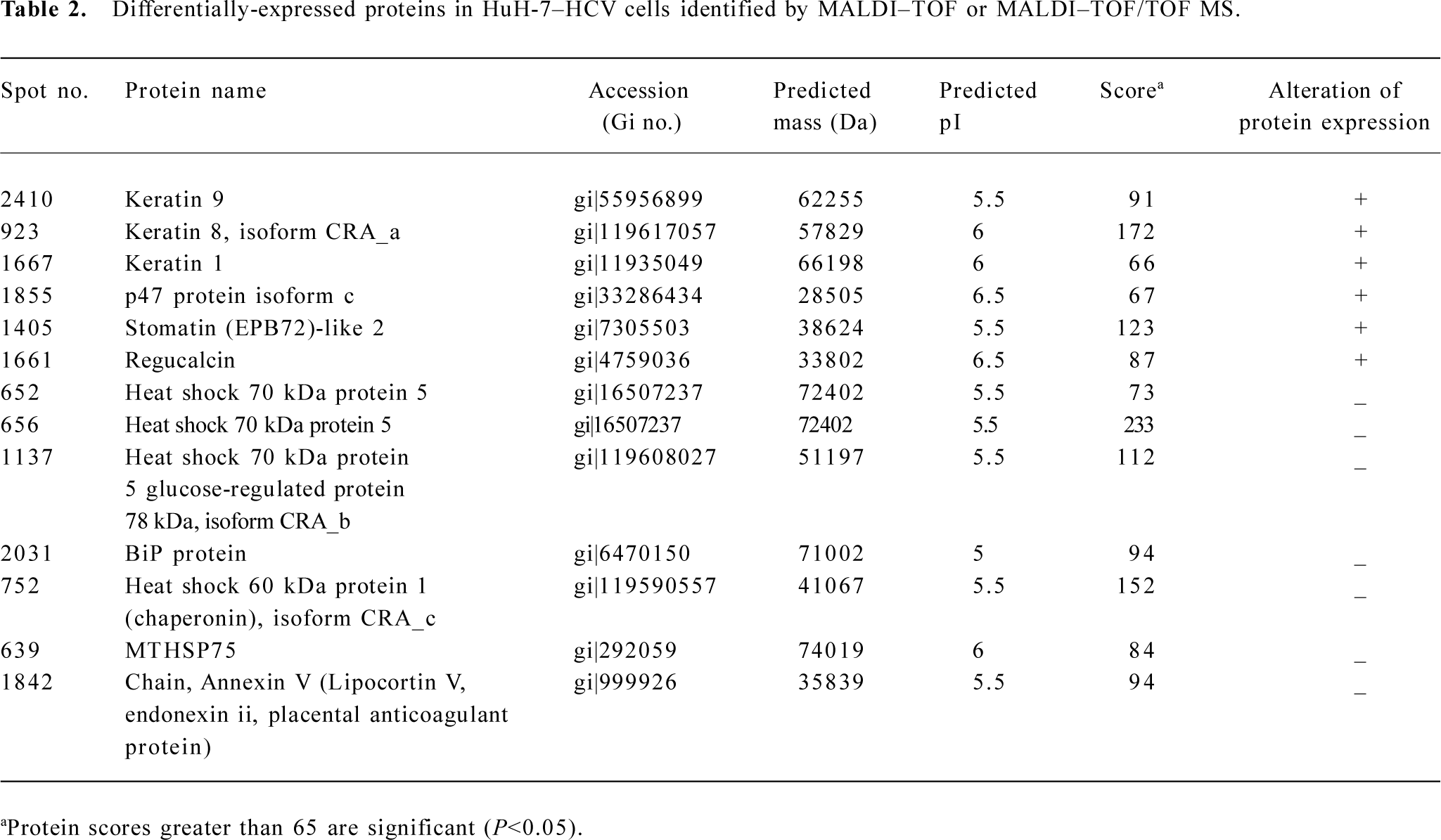
The spots of interest were excised from the 2D gels and subjected to MALDI–TOF MS or MALDI–TOF/TOF MS. The resulting PMF data were used to search the NCBI databases via MASCOT software. The identity of specific proteins was determined by comprehensively comparing the corresponding experimental pI, Mr, the number of matched peptides, and the sequence coverage. Upregulated proteins in HuH-7–HCV cells included keratin 9, keratin 8 isoform CRA_a, keratin 1, p47 protein isoform c, stomatin (EPB72)-like 2 (SLP-2), and regucalcin. Downregulated proteins included heat shock 70 kDa protein 5, heat shock 70 kDa protein 5 glucose-regulated protein 78 kDa (GRP 78) isoform CRA_b, heat shock 60 kDa protein 1 isoform CRA_c, MTHSP75, and Annexin V (Table 2).
Full table
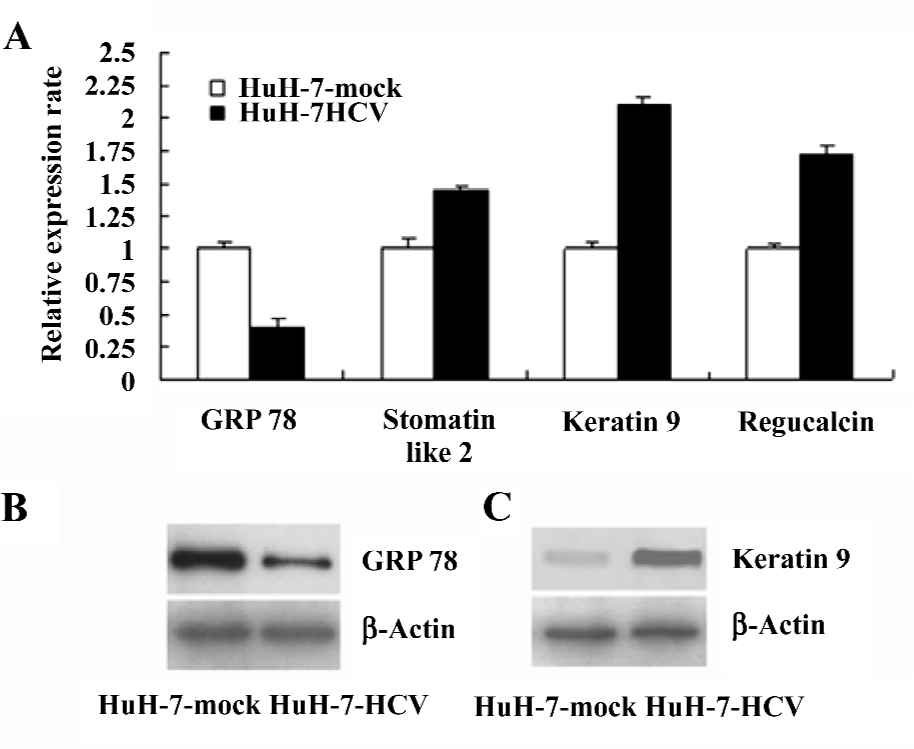
Verification of specific differentially-expressed proteins by real-time RT–PCR and Western blotting The differential expression of the genes encoding GRP 78, stomatin-like 2, keratin 9, and regucalcin were validated by real-time RT–PCR (Figure 3A). The differential expression of the GRP 78 (Figure 3B) and keratin 9 (Figure 3C) proteins was verified by Western blotting. The changes in the expression levels of these selected proteins were consistent with the 2DE results. Such results demonstrate that the proteomic analysis of HuH-7–HCV and HuH-7 mock cells can reliably identify cellular proteins that are differentially expressed as the result of the replication and expression of the HCV genome.
Discussion
In order to establish protein expression profiles and to characterize HCV infection-related proteins, HuH-7 cells were transfected with in vitro-transcribed HCV full length RNA. The differential expression proteins of HuH-7 harboring HCV-RNA and HuH-7 mock cells were analyzed by 2DE and MS. The identified differentially-expressed proteins included proteins associated with the host cell cytoskeleton, intracellular trafficking, molecular chaperones, apoptosis, protein degradation, oxidative and endoplasmic reticulum (ER) stress, cell cycle progression, antigen presentation, and calcium homeostasis.
Several members of the keratin family (keratin 1, 9, and 8) were upregulated after HCV-RNA transfection. Keratins are proteins that form the intermediate filament cytoskeleton and are essential for normal tissue structure and function. Keratin 8 is a type II keratin and occurs in many epithelial cells, including hepatocytes. Several isoforms of keratin 8 generated by differential splicing have been identified. The majority of keratin 8 is assembled with its partner, keratin 18, into highly insoluble 10 nm filaments that extend from the nucleus to the internal leaflet of the plasma membrane[16]. The function of keratin 8/18 in the liver is protection from mechanical and non-mechanical forms of stress[17], which is supported by the association of several K8/18 mutations with cryptogenic and non-cryptogenic forms of human liver disease[18]. It has been documented that keratin 8/18 has been linked to tumor necrosis factor-induced and Fas-mediated apoptosis[19]. There is increasing evidence suggesting that liver cell damage in chronic HCV infection is mediated by the induction of apoptosis[20]. Engulfment of apoptotic bodies by hepatic stellate cells stimulates the fibrogenic activity of these cells and may be one mechanism by which hepatocyte apoptosis promotes fibrosis[21]. Thus our findings indicated that the upregulation of keratin 8 agreed with this proposed mecha-nism.
HCV utilizes the host cell ER as the main site for replication and protein synthesis. Recent studies have been demonstrated that HCV proteins have the propensity to induce distinct alterations of ER membranes[22] and homeostasis, and cause some level of ER stress. The ER stress response works at the level of protein translation and appears to be regulated by the level of stress. Mild ER stress inhibits protein synthesis initiation and can slow cell growth, while prolonged ER stress can lead to apoptosis. These events correlate with the transcriptional induction of the ER chaperone GRP 78, a typical marker of ER stress. Heat shock 70 kDa protein 5 GRP 78 is also known as heat shock 70 kDa protein 5 and immunoglobulin binding protein (BiP). It belongs to the Hsp70 multigene family. In humans, the Hsp70 multigene family consists of at least 4 members: Hsp70, GRP 78, MTHSP75, and Hsc70. The molecular chaperone GRP 78 is important in the folding, maturation, and transport of proteins out of the cell, and GRP 78 is critical for the unfolded protein response required to alleviate ER “stress”, maintain ER function, and protect cells against death[23]. It works as a coreceptor for viruses and MHC I antigen presentation, and as a receptor for activated forms of the plasma proteinase inhibitor α2-macroglobulin[24]. GRP 78 is also identified as a keratin-associated protein. The GRP 78–keratin 8/18 association is induced by tunicamycin or glucose starvation. The physiological relevance of keratin 8/18–GRP 78 binding remains to be determined. The location of GRP 78 in the ER makes any association of K8/18 with GRP 78 significant only under conditions of ER damage, or export of resident luminal ER proteins into the cytosol or the cytoplasmic resident soluble keratins into the ER[25]. The synthesis of HCV proteins may influence cell fate through the regulation of the ER stress signaling pathway and apoptotic response.
A mitochondrial chaperone, heat shock 60 kDa protein, was downregulated in the HuH-7–HCV cells. This chaperone may protect mitochondria from oxidative stress by facilitating the proper assembly of mitochondrial proteins[26]. The downregulation of this chaperone has been reported in various tumor series correlating with disease outcome[27]. Acute ablation of Hsp60 results in destabilization of survivin levels, and nearly complete and selective loss of the mitochondrial pool of survivin[28], thus abrogating this anti-apoptotic response[29]. MTHSP75 is another mitochondrial chaperone for the import and folding of newly-synthesized nuclear and mitochondrial-encoded proteins[30]. It has been reported to be involved in antigen processing and presentation[31]. Therefore, the downregulation of MTHSP75 may contribute to the cytopathic effect on mitochondria induced by HCV and possibly impairs antigen presentation.
There is no previous report concerning the relationship between HCV infection and the SLP-2, regucalcin, Annexin V, and p47 proteins. SLP-2 is a novel stomatin homolog and is mainly localized in the cytoplasm with some distribution on the membrane[32]. SLP-2 is a cancer-related gene whose product promotes cell growth, tumorigenicity, and adhesion in human esophageal squamous cell carcinoma, endometrial adenocarcinoma, and laryngeal squamous cell carcinoma. Furthermore, the SLP-2 expression correlates with the clinical stage of carcinoma. Zhang et al showed that SLP-2 was overexpressed in all the tumor types they tested compared with their normal counterparts[33]. The etiology of the increased SLP-2 expression in human cancers is unknown. Possible mechanisms include point mutation, gene amplification, gene rearrangement, and insertion of a strong promoter or enhancer. Epigenetic modifications, including demethylation and deacetylation may also be responsible.
Regucalcin (senescence marker protein-30) is a multifunctional protein that is expressed in multiple tissues, including hepatocytes and renal tubular epithelia[34]. Regucalcin regulates calcium homeostasis for the maintenance of hepatocyte morphology and differentiation, inhibits cell death, and provides protection againt cell injury and oxidative stress by enhancement of Ca2+ efflux to either the extracellular space or intraorganellar spaces through membrane Ca2+ pump activity[35]. In the present study, the upregulation of regucalcin may be the response of the cell to the injury caused by HCV.
Annexin V is a member of the Annexin family of structurally-homologous proteins interacting with anionic phospholipid membranes in a reversible calcium-dependent manner[36]. It was shown to interact with membrane proteins and to regulate the sodium–calcium exchanger[37]. In a previous study, Annexin V was abundant in normal mucosa compared with neighboring tumor tissue[38]. P47 is an adaptor molecule of the cytosolic ATPase p97. The principal role of the p97–p47 complex is known to be involved in the heterotypic fusion of transport vesicles with their target membranes, the homotypic fusion of membrane compartments and ubiquitin-dependent protein degradation[39]. In the present study, the level of the p47 protein was higher than the control and it is worth further study to elucidate the role of p47 in HCV replication.
In summary, the progression of HCV infection involves various cellular and molecular mechanisms. These results provide a further understanding of the molecular mechanism of cellular events and pathogenesis associated with viral progression, and are useful for the identification of HCV infection-associated proteins which will be molecular targets for treatment.
Author contribution
Meng XUN, Si-hai ZHAO, Yong-lie CHU designed research; Meng XUN, Si-hai ZHAO, Chun-xia CAO, Juan SONG performed research; Meng XUN, Si-hai ZHAO, Yong-lie CHU contributed new reagents or analytic tools; Meng XUN, Si-hai ZHAO, Ming-ming SHAO analyzed data; Meng XUN, Juan SONG, Yong-lie CHU wrote the paper.
Acknowledgement
We would like to thank Shaanxi Hepatopathy Institute, China, for enabling us to use the Biosafety level 2 laboratory to perform this study. We gratefully thank Tong Zhu, Yi-hua Wang, and Li-peng Yan (Shaanxi Hepatopathy Institute) for their guidance and technical assistance. Our thanks also to William T Ruyechan (University at Buffalo, SUNY) for editing the English version of this manuscript.
References
- Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexander WJ, et al. The sentinel counties chronic non-A NBHST: the natural history of community-acquired hepatitis C in the United States. N Engl J Med 1992;327:1899-905.
- Choo Q, Richman KH, Han JH, Berger K, Lee C, Dong C, et al. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci USA 1991;88:2451-5.
- Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol 2000;242:55-84.
- Hijikata M, Kato N, Ootsuyama Y, Nakagawa M, Shimotohno K. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc Natl Acad Sci USA 1991;88:5547-51.
- Blight KJ, Rice CM. Secondary structure determination of the conserved 98-base sequence at the terminus of hepatitis c virus genome RNA. J Virol 1997;71:7345-52.
- Kolykhalov AA, Feinstone SM, Rice CM. Identification of a highly conserved sequence element at the 3´ terminus of hepatitis C virus genome RNA. J Virol 1996;70:3363-71.
- Yanagi M, St Claire CM, Emerson SU, Purcell RH, Bukh J. In vivo analysis of the 39 untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc Natl Acad Sci USA 1991;96:2291-5.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000;81:1631-48.
- Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999;285:110-3.
- Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J Virol 2003;77:3181-90.
- Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol 2002;76:2997-3006.
- Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, Rutter G, et al. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J Virol 2002;76:4008-21.
- Tang HL, Chu YL, Zhang SL, Guo WX, inventors; Yangling, Daiying Biolog Engine (CN), assignee. The intact hepatitis C virus and the method for culturing it in cell culture. European Patent 1 424 390. 2004 Feb 06.
- Yan JX, Wait R, Berkelman T, Harry RA, Westbrook JA, Wheeler CH, et al. A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis 2000;21:3666-72.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-8.
- Gonias SL, Hembrough TA, Sankovic M. Cytokeratin 8 functions as a major plasminogen receptor in select epithelial and carcinoma cells. Front Biosci 2001;1:1403-11.
- Omary MB, Ku NO, Toivola DM. Keratins: guardians of the liver. Hepatology 2002;35:251-7.
- Ku NO, Darling JM, Krams SM, Esquivel CO, Keeffe EB, Sibley RK, et al. Keratin 8 and 18 mutations are risk factors for developing liver disease of multiple etiologies. Proc Natl Acad Sci USA 2003;100:6063-8.
- Sorom AJ, Nyberg SL, Gores GJ. Keratin, fas, and cryptogenic liver failure. Liver Transpl 2002;8:1195-7.
- Bantel H, Schulze-Osthoff K. Apoptosis in hepatitis C virus infection. Cell Death Differ 2003;10:48-58.
- Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology 2004;39:273-8.
- Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 2002;76:5974-84.
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res 2005;569:29-63.
- Misra UK, Gonzalez-Gronow M, Gawdi G, Pizzo SV. The Role of MTJ-1 in cell surface translocation of GRP78, a receptor for α2-macroglobulin-dependent signaling. J Immunol 2005;174:2092-7.
- Liao J, Price D, Omary MB. Association of glucose-regulated protein (grp78) with human keratin 8. FEBS Lett 1997;417:316-20.
- Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, et al. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid betapeptide (1–42) into rat brain: implications for Alzheimer’s disease. Neuroscience 2005;132:313-24.
- Cappello F, Di Stefano A, David S, Rappa F, Anzalone R, La Rocca G, et al. Hsp60 and Hsp10 down-regulation predicts bronchial epithelial carcinogenesis in smokers with chronic obstructive pulmonary disease. Cancer 2006;15:2417-24.
- Caldas H, Jiang Y, Holloway MP, Fangusaro J, Mahotka C, Conway EM, et al. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 2005;24:1994-2007.
- Dohi T, Xia F, Altieri DC. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell 2007;27:17-28.
- Mizzen LA, Kabiling AN, Welch WJ. The two mammalian mitochondrial stress proteins, grp 75 and hsp 58, transiently interact with newly synthesized mitochondrial proteins. Cell Regul 1991;2:165-79.
- Manara GC, Sansoni P, Badiali-De Giorgi L, Gallinella G, Ferrari C, Brianti V, et al. New insights suggesting a possible role of heat shock protein 70-kD familyrelated protein in antigen processing/presentation phenomenon in humans. Blood 1993;82:2865-71.
- Sprenger RR, Speijer D, Back JW, De Koster CG, Pannekoek H, Horrevoets AJ. Comparative proteomics of human endothelial cell caveolae and raftsusing two-dimensional gel electrophoresis and mass spectrometry. Electrophoresis 2004;25:156-72.
- Zhang L, Ding F, Cao W, Liu Z, Liu W, Yu Z, et al. Stomatin-like protein 2 is overexpressed in cancer and involved in regulating cell growth and cell adhesion in human esophageal squamous cell carcinoma. Clin Cancer Res 2006;12:1639-46.
- Mori T, Ishigami A, Seyama K, Onai R, Kubo S, Shimizu K, et al. Senescence marker protein-30 knockout mouse as a novel murine model of senile lung. Pathol Int 2004;54:167-73.
- Sar P, Rath B, Subudhi U, Chainy GB, Supakar PC. Alterations in expression of senescence marker protein-30 gene by 3,3´,5-triiodo-L-thyronine (T3). Mol Cell Biochem 2007;303:239-42.
- Raynal P, Pollard HB. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium and phospholipid-binding proteins. Biochim Biophys Acta 1994;1197:63-93.
- Camors E, Charue D, Trouvé P, Monceau V, Loyer X, Russo-Marie F, et al. Association of annexin A5 with Na+/Ca2+ exchanger and caveolin3 in non-failing and failing human heart. J Mol Cell Cardiol 2005;40:47-55.
- Friedman DB, Hill S, Keller JW, Merchant NB, Levy SE, Coffey RJ, et al. Proteome analysis of human colon cancer by two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics 2004;4:793-811.
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, et al. p47 is a cofactor for p97-mediated membrane fusion. Nature 1997;388:75-8.