
Effect of β2-adrenergic agonist clenbuterol on ischemia/reperfusion injury in isolated rat hearts and cardiomyocyte apoptosis induced by hydrogen peroxide
Introduction
Previous studies have shown that cardiomyocyte ischemia/reperfusion (I/R) injury could lead to cardiac dysfunction, such as arrhythmias, tachycardia, ventricular fibrillation, and subsequent cell death[1]. The mechanisms of I/R injury include oxidative stress, apoptosis, and intracellular calcium overload[2-4].
Clenbuterol is a β2-adrenergic receptor (AR) agonist, which was first used in the mid 1970s to treat asthma, and has been approved for this indication in Europe. Recently, clenbuterol has been regarded as a potential treatment for cardiac diseases, specifically in the patients who underwent implantation of left ventricular assist device and had dilated cardiomyopathy[5-7]. Experimental results have provided evidence that clenbuterol could decrease apoptosis and improve calcium homeostasis in a rodent model of ischemic cardiomyopathy[8]. A recent study also found that clenbuterol could elevate Bcl-2 and Bcl-xl protein levels[9] and induce growth factor mRNA in the rat hippocampus after transient forebrain ischemia[10-12]. Moreover, evidence showed that the toxic effects of β-adrenergic stimulation was mediated primarily via β1 receptors, whereas β2-receptor stimulation may be protective[13-16]. These findings suggest that clenbuterol may alleviate myocardial ischemia (MI)/R injury. However, the effect of clenbuterol on MI/R injury has not been evaluated to date in any experimental or clinical study.
Therefore, we designed the experiment to observe the effect of clenbuterol on MI/R injury, including apoptosis, oxidative stress, and Ca2+-ATPase activity in isolated rat hearts. Meanwhile, cultured newborn rat cardiomyocytes were exposed to exogenous hydrogen peroxide (H2O2) to determine the effects of clenbuterol on oxidative stress-induced cardiomyocyte apoptosis and injury in vitro.
Materials and methods
Animals Adult male Wistar rats (220±30 g) were provided by the Experimental Animal Center of Tongji Medical College (Wuhan, China). All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health.
Drugs Clenbuterol was obtained from Sigma (St Louis, MO, USA). ICI118551{(±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride} was purchased from Zeneca Pharmaceuticals (Cheshire, UK). All compounds were diluted in standard Krebs–Henseleit buffer (KHB), containing (in mmol/L): 119 NaCl, 4 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, 25 NaHCO3, and 10 glucose at pH 7.4.
Isolated rat heart preparation and experimental protocol The rats were heparinized (500 U/kg, ip) and then anesthetized with 20% pentobarbital sodium (50 mg/kg, ip). The rat hearts were excised rapidly and the aortas were cannulated. The hearts were mounted in a Langendorff apparatus and perfused with KHB, which was equilibrated with 95% O2 and 5% CO2 at 37 °C.
Forty rats were randomly divided into 4 experimental groups: (1) control (N) group (n=10) was only perfused with KHB for 110 min; (2) I/R group (n=10) underwent 20 min of perfusion, 30 min of no-flow global ischemia, and 60 min of reperfusion; (2) clenbuterol (CLEN) group (n=10) received 1 µmol/L clenbuterol in KHB solution 10 min before global ischemia and through reperfusion (60 min); and (4) ICI118551 (ICI) group (n=10), where ICI was used as the β2-AR antagonist to pretreat the hearts for 10 min with 30 nmol/L before clenbuterol treatment, and then served as the CLEN group.
A previous study demonstrated that clenbuterol at a dose of 1 µmol/L can induce β2-AR activity and 30 nmol/L ICI118551 can effectively inhibit β2-AR activity[6,17,18].
The cardiac parameters of heart rate (HR), left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), left ventricular developed pressure (LVDP, LVDP=LVSP-LVEDP), and maximal rise rate of left ventricular pressure (+dp/dtmax) were monitored continuously. During the experiment, the coronary effluent (CF) was collected as an index of vascular diastolic function.
Thirty minutes after reperfusion, the CF was collected for the lactate dehydrogenase (LDH) assay. At the end of the reperfusion, the left ventricle was quickly removed and stored at -80 °C for different assays.
Histomorphological analysis After the measurement of hemodynamics, the hearts were quickly removed and cut into transverse slices from apex to base and processed for histology. Subsequently, the hearts were fixed in a 10% formalin solution for 24 h, embedded in paraffin wax, and serially sectioned at 7 µm by routine protocols[19].
For the histomorphological analysis, the sections were stained with hematoxylin–eosin (HE) and examined using a light microscope (Olympus BX40, Tokyo, Japan).
Measurement of LDH in the coronary effluent, ma-londialdehyde, and superoxide dismutase in cardiac tissue The frozen cardiac tissue samples were weighed and homogenized (1:10, w/v) in 50 mmol/L phosphate buffer (pH 7.4) and kept in an ice-bath. The malondialdehyde (MDA) level and superoxide dismutase (SOD) activity were measured by colorimetric analysis using a spectrophotometer with the associated detection kits (Jiancheng, Nanjing, China), respectively.
LDH in the CF was determined by an automatic biochemistry analyzer (Toshiba model 750, Tokyo, Japan) using ultraviolet detection[20]. The LDH, MDA, and SOD levels are expressed as IU/L, nmol/mg protein, and U/mg protein, respectively.
Preparation of sarcoplasmic reticulum from rat heart Sarcoplasmic reticulum (SR) was prepared according to the methods of Kodavanti et al[21-23]. The hearts were transferred to ice-cold homogenizing medium containing (in mmol/L): 50 Na2HPO4, 10 Na2EDTA, and 25 NaF at pH 7.4. Briefly, the minced ventricular tissue were homogenized in 10 mL of ice-cold homogenizing medium. An additional 5 mL homogenizing medium was added and the homogenate was sedimented twice for 20 min at 14 000×g at 4 °C. The supernatant was recentrifuged at 45 000×g for 30 min. The pellet obtained after this centrifugation, consisting of crude membrane vesicles (SR), was suspended in storage buffer consisting of 30 mmol/L histidine, 0.25 mol/L sucrose, 10 mmol/L EDTA, and 10 mmol/L NaF at pH 7.4 to a final concentration of 30–40 mg/mL and stored at –80 °C until used.
Measurement of Ca2+-ATPase activity The SR Ca2+-ATPase activity was determined colorimetrically by measuring the inorganic phosphate (Pi) liberated from ATP hydrolysis with a kit (Jiancheng, China)[22]. Ca2+-ATPase activity was assayed in a medium containing 50 mmol/L histidine (pH 7.0), 3 mmol/L MgCl2, 0.1 mol/L KCl, 5 mmol/L sodium azide, 3 mmol/L ATP, and 0.05 mmol/L CaCl2. Cardiac SR membranes were added to the reaction mixture at a final concentration of 15–20 µg/mL and pre-incubated for 10 min at 37 °C. The reaction was initiated by the addition of ATP. Ca2+-ATPase activity was the activity that was inhibited by 1 mmol/L egtazic acid (EGTA). Mitochondrial contamination was inhibited by 5 mmol/L sodium azide[24]. The Ca2+-ATPase activity was expressed as µmol Pi/mg protein per hour in this study.
In situ apoptosis assay An in situ apoptosis detection kit (Dead End colorimetric TUNEL System, Promega) was used to detect apoptosis. As described previously, the sections were deparaffinized in xylene, rehydrated, incubated with 20 mg/mL proteinase K for 10 min, and rinsed in distilled water. Endogenous peroxidase activity was inhibited by incubation with 3% H2O2 for 5 min. The sections were then incubated with equilibration buffer for 10–15 s and TdT enzyme, which was prepared according to the kit instructions, in a humidified atmosphere, at 37 °C for 60 min. They were subsequently placed in prewarmed working strength stop/wash buffer at room temperature for 10 min and incubated with antistreptavidin peroxidase, at a 1:500 dilution in phosphate-buffered saline (PBS) for 45 min. Each step was separated by careful washing in PBS. Labeling was revealed using 3,3´-diaminobenzidine-tetrachloride/H2O2, and nuclei were counterstained with Mayer’s hematoxylin. Four slide fields were randomly examined with a 200×magnification.
RT–PCR for Bcl-2 and Bax mRNA Total RNA was extracted from the left ventricular myocardium using Trizol reagent (Gibco BRL, USA). First-strand cDNA was reversed transcribed using oligo-dT. Two microliters of cDNA were amplified by PCR using the following primers: Bcl-2, 5'-CATCTTCTCCTTCCAGCC-3' and 5'-GGAGGAAATCAACAGACGTCGC-3';Bax-α, 5'-TCGTGATGGCAACTT-CAACT-3' and 5'-GTGAGGACTCCAGCCACAAA-3'; and β-actin, 5'-TGGAATCCTGTGGCATCCATGAAAC-3' and 5'-TAAAACGCAGCTCAGTAACAGTCCG-3'. The PCR cycling program was set for 1 cycle of predenaturation at 94 °C for 4 min and then 25 cycles at 94 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s, followed by 1 cycle at 72 °C for 10 min. PCR products were visualized by UV illumination after electrophoresis on a 2% agarose gel. The gel photographs were scanned with a computerized densitometer (SYNGENE, London, UK).
Western blot analysis of caspase-3 proteins For Western blot analysis, protein extracts were prepared from the left ventricular myocardium using extraction buffer (20 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 3% SDS, 1 mmol/L phenylmethylsulfonyl fluoride, and 5 µg/mL protease inhibitor cocktail). In total, 50 µg protein samples were loaded onto 10% SDS–PAGE. Following electrophoresis, the proteins were transferred to a nitrocellulose transfer membrane. After incubation in blocking buffer (5% non-fat dry milk in TBST) for 1 h, rat monoclonal caspase-3 primary antibodies (Cell Signaling Technology) were added to the membrane at 1:1000 dilution in TBST and incubated at 4 °C overnight. After washing the membrane, the blots were incubated with a horseradish peroxidase-labeled secondary antibody. Immunoreactive bands were visualized with an ECL reagent.
Cell culture and treatment Neonatal ventricular myo-cytes were prepared from 1–4-day-old Wistar rats under sterile conditions. After the removal of the blood vessels and atria, the ventricle was put into cold D-Hanks’ solution, minced and incubated in 0.2% trypsin at 37 °C for 10 min. The supernate was discarded and the tissue was redigested with trypsin for 10 min; equal Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum was then used to terminate digestion, and the supernate was centrifuged at 700×g for 10 min. The sediment was resuspended in DMEM medium containing 10% fetal bovine serum. The residual tissues were redigested for 4–6 cycles until a single cell was obtained. Cell suspensions were collected together and plated in culture flasks for 60 min in a humidified atmosphere of 5% CO2 at 37 °C to remove fibroblasts. The unattached cells were counted and seeded into another culture flask at a density of 1.5×106/L. The medium was changed every 48 h. All the cells were cultured for 3–5 d when synchronously contracting.
The myocytes were exposed to one of the following treatments: (1) control: receiving no drugs; (2) clenbuterol: receiving 1 µmol/L for 24 h; (3) H2O2: receiving 100 µmol/L H2O2 for 24 h; (4) CLEN+H2O2: pretreated with 1 µmol/L clenbuterol for 10 min before 100 µmol/L H2O2 for 24 h; and (5) ICI+CLEN+H2O2 group: pretreated with 30 nmol/L ICI118551 for 10 min; then served as the CLEN+H2O2 group.
Cell viability analysis Cell viability was assessed by measuring the mitochondrial dependent reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan. Cells were dispensed into 96 well plates at 1×105 cells/mL in 100 µL volumes. After the treatment, 10 µL MTT solution was added to each well giving a final concentration of 0.5 mg/mL. After incubation for 4 h at 37 °C, 100 µL DMSO was added to dissolve the formazan crystals. Absorbance was read at 570 nm, which was measured by a DG3002 ELISA plate reader (HuaDong Electronic, Nanjing, China).
Flow cytometry analysis For the flow cytometry analysis, the cells (>1×106) were digested with 0.2% trypsin and collected by centrifugation. After washing twice with ice-cold PBS, the cells were added 10 µL Annexin V–fluorescein-isothiocyanate and 5 µL propidium iodide to an 85 µL cell suspension. After incubation for 15 min in the dark, stained cells were analyzed via flow cytometry using a FACSCalibur system with CellQuest software (BD Pharmingen).
Statistical analysis The results are shown with mean±SEM. Data were analyzed with SPSS version 11.5 software (SPSS, Chicago, IL, USA). Any difference in each parameter among groups was evaluated with one-way ANOVA with the Tukey–Kramer multiple comparisons test. P<0.05 was considered statistical significance.
Results
Effect of clenbuterol on myocardial functions after MI/R Pretreatment with clenbuterol decreased the LVEDP and increased the CF compared with the I/R groups at 20, 30, 40, 50, and 60 min of reperfusion (P<0.05; Figure 1B,1D). The ICI118551+clenbuterol group had no difference compared with the I/R group.
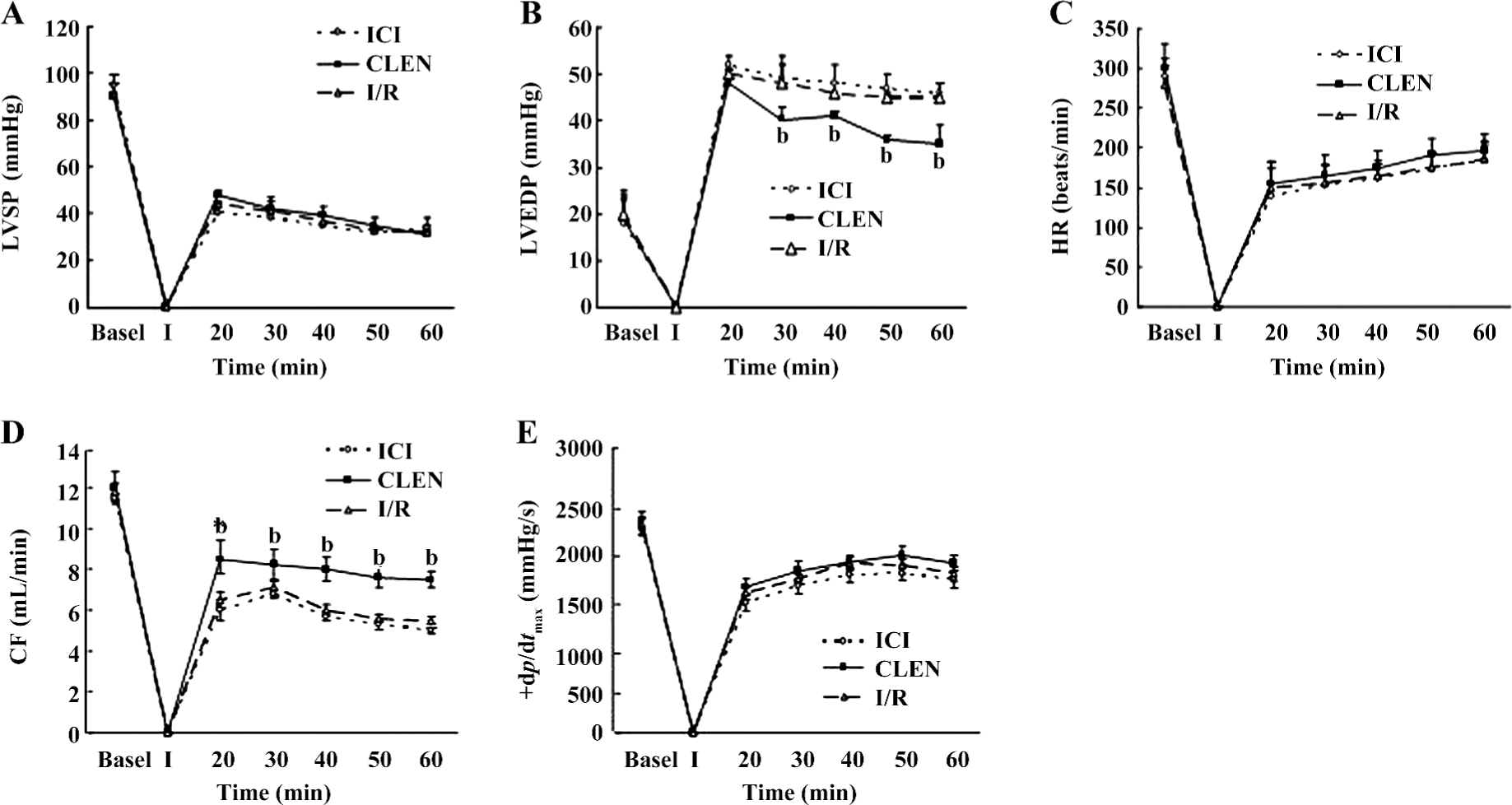
However, the LVSP, +dp/dtmax, and HR were not improved by clenbuterol treatment (Figure 1A,1C,1E). There was no difference among the 3 groups.
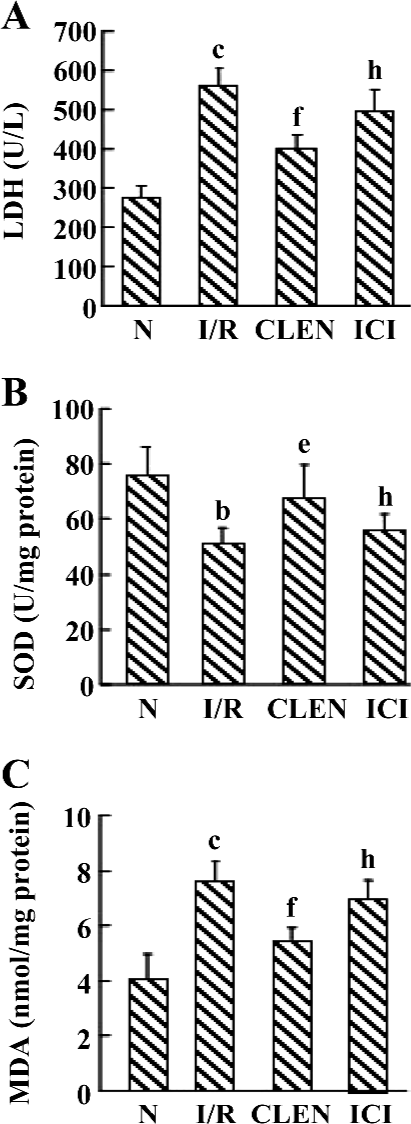
Effect of clenbuterol on LDH, MDA, and SOD levels after MI/R Data are shown in Figure 2. Compared with the controls, the I/R-induced injury was manifested by a significantly increased MDA content and LDH release while SOD activity significantly decreased. The administration of clenbuterol caused a significant decrease in the MDA content and LDH release and increased in SOD enzyme activity compared with the I/R group (P<0.01). Moreover, pretreatment of ICI118551 led to an increase in MDA and LDH and a decrease in SOD compared with the clenbuterol group (P<0.05).
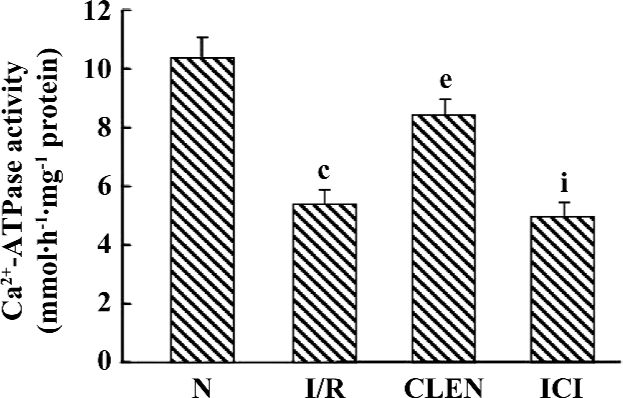
Effect of clenbuterol on Ca2+-ATPase activity The Ca2+-ATPase of SR catalyzes the most important step in relaxation by coupling cleavage of ATP to transport 2 Ca2+ into the SR lumen. To study whether Ca2+-ATPase is involved in the clenbuterol-induced improvement of diastolic function following I/R, the present study examined Ca2+-ATPase activity by an optical assay. As shown in Figure 3, the SR Ca2+-ATPase activity was significantly decreased in the I/R group compared with that of the control group (5.421±0.495 µmolPi·h–1·mg–1 protein vs 10.369±0.724 µmolPi·h–1·mg–1 protein, P<0.01). Interestingly, treatment with clenbuterol significantly increased the SR Ca2+-ATPase activity compared with the I/R group (8.452±0.517 µmolPi·h–1·mg–1 protein), whereas the pretreatment of ICI118551 decreased the SR Ca2+-ATPase activity compared with the clenbuterol group (4.939±0.547 µmolPi·h–1·mg–1 protein). The results suggested that increased Ca2+-ATPase activity might be involved in the clenbuterol-induced improvement diastolic function following MI/R.
Histological study Representative slides from each group are shown in Figure 4A. After I/R, prominent interstitial edema was present on HE-stained tissue sections, whereas nearly no edema was demonstrated in the clenbuterol groups (Figure 4A). The degree of edema in the ICI118551 group was increased compared with the clen-buterol group (Figure 4A).
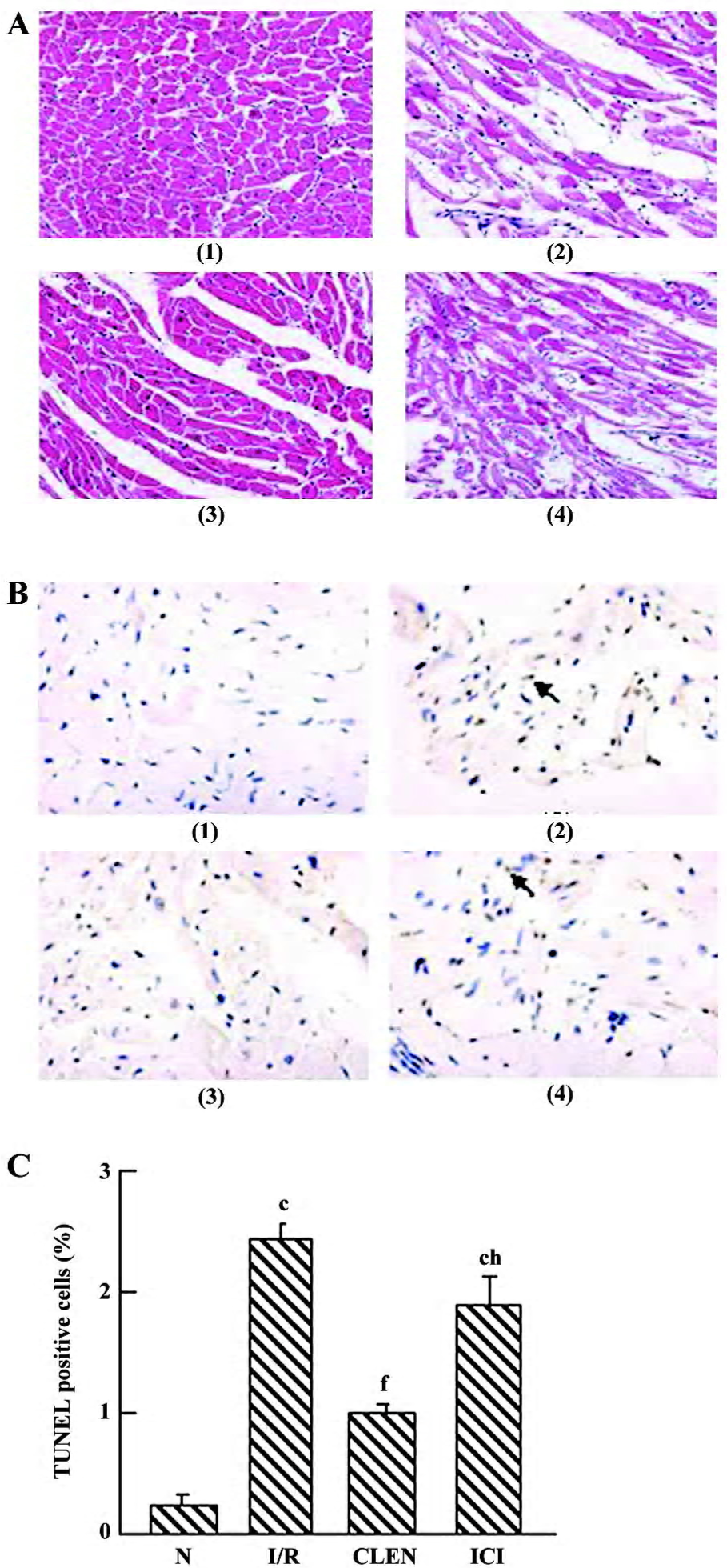
Effect of clenbuterol on apoptosis We detected single TUNEL-positive cells in the myocardium (Figure 4B,4C). The number of terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick-end labeling (TUNEL)-positive cells increased significantly in the I/R group compared with the control group (P<0.01). The treatment of clenbuterol decreased TUNEL-positive cells compared with the I/R group (P<0.01). Moreover, the pretreatment of ICI118551 enhanced TUNEL-positive cells compared with the clenbuterol group (P<0.01).
Effect of clenbuterol on Bcl-2 and Bax expression As shown in Figure 5, clenbuterol significantly increased Bcl-2 and decreased Bax expressions compared with the I/R group. ICI118551 revealed a decrease of Bcl-2 and an increase of Bax mRNA levels compared with the clenbuterol groups. These results suggested that clenbuterol inhibited myocardial apoptosis by decreasing the ratio of Bax/Bcl-2 mRNA after MI/R.
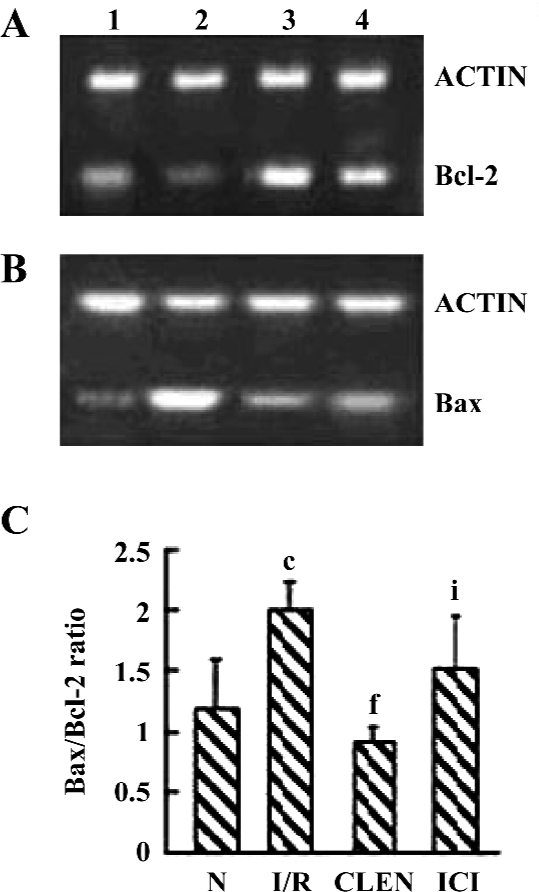
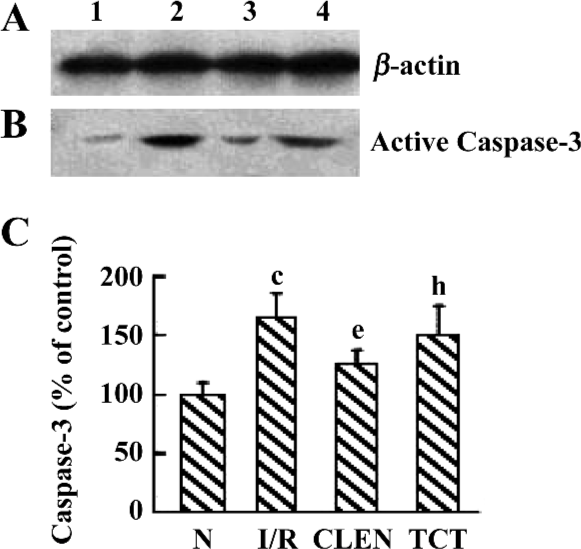
Effect of clenbuterol on caspase-3 protein expression We investigated the active p17 subunit of caspase-3, which indicated the proteolytic activation of caspase-3. As illustrated in Figure 6, an obvious increase of p17 was detectable after myocardial I/R compared with the control group. Clenbuterol reduced the caspase-3 expression compared with the I/R groups. However, ICI118551 revealed enhanced caspase-3 activity compared with the clenbuterol group. These results suggested that clenbuterol decreased the caspase-3 expression to inhibit myocardial apoptosis after MI/R.
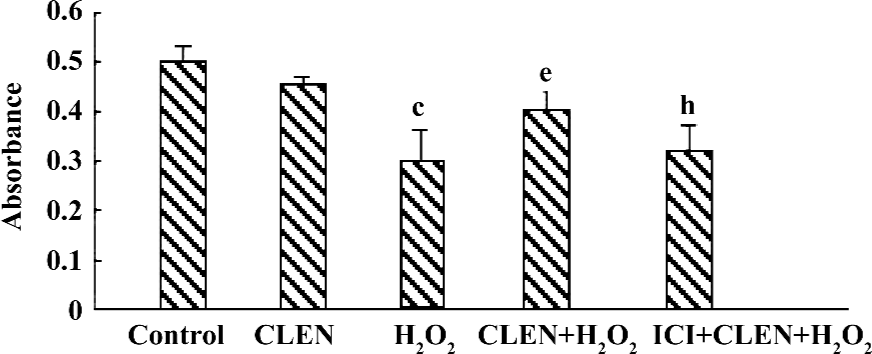
Effect of clenbuterol on cell viability in H2O2-induced injury The exposure of cardiomyocytes to 100 µmol/L H2O2 for 24 h produced an obvious decrease in cell viability as measured by MTT (P<0.01 vs the control group). When the cells were pretreated with clenbuterol, cell viability showed no difference to the control group. However, the CLEN+H2O2 group had increased cell viability compared with the H2O2 group (P<0.01). The ICI+CLEN+H2O2 group had an adverse effect compared with the CLEN+H2O2 group (P<0.05; Figure 7).
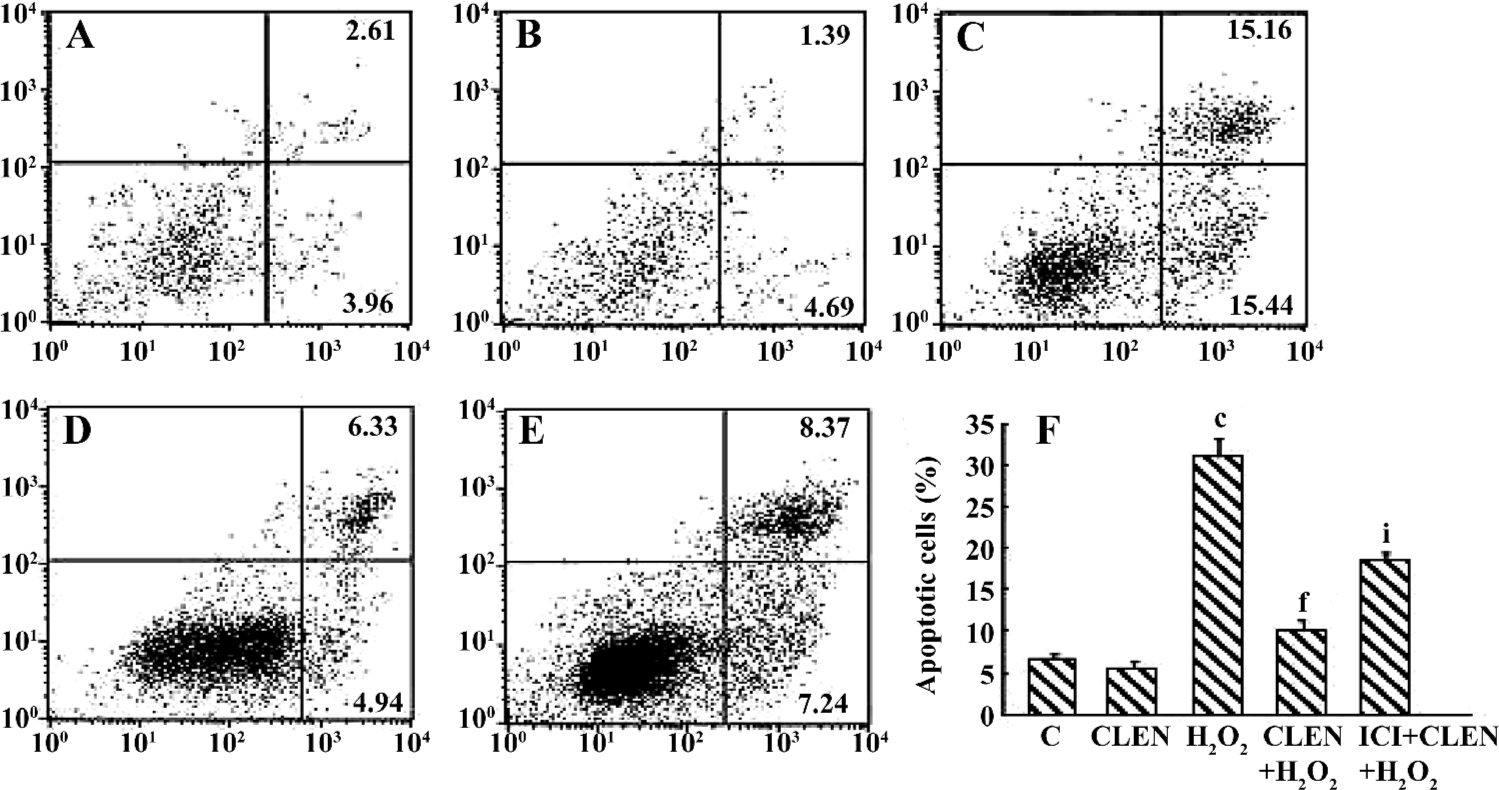
Effect of clenbuterol on H2O2-induced cardiomyocyte apoptosis When exposed to 100 µmol/L H2O2 for 24 h, the percentage of apoptotic cells increased from 6.5%±0.7% in the control group to 30.9%±2.3% in the H2O2 group (P<0.01). In the CLEN+H2O2 group the number of apoptotic cells decreased compared with the H2O2 group (10.23%±1.2% vs 30.9%±2.3%, P<0.01). In the ICI+CLEN+H2O2 group the apoptotic rate increased to 18.56%±1.3% compared with the CLEN+H2O2 group (Figure 8).
Discussion
In the present study, we found that clenbuterol significantly improved diastolic function (LVEDP and CF) in the MI/R isolated rat hearts, which is partly due to increased Ca2+-ATPase activity. Clenbuterol significantly increased SOD activity and decreased the MDA level and LDH release after MI/R. Furthermore, clenbuterol decreased apoptosis and inhibited caspase-3 protein and Bax/bcl-2 mRNA expressions. Clenbuterol also reduced cardiomyocyte apoptosis induced by H2O2 in vitro. These findings suggest that clenbuterol plays an important role in the cardiac protection of MI/R injury.
It has been well established that both β1- and β2-AR are linked to the Gs-protein and potentially activate protein kinase A; however, β2-AR can also couple to Gi–PI3K–Akt signaling pathways to protect cardiomyocytes. As β2-AR stimulation attenuates left ventricular remodeling and decreases apoptosis in a rodent model of ischemic cardiomyopathy[8], we confer that clenbuterol has beneficial effect on MI/R injury.
In our study, we found that pretreatment of clenbuterol improved left ventricular diastolic function including LVEDP and CF and decreased LDH release. These findings supported those of a previous report by Ahmet et al[25], who used the β2-adrenergic agonists fenoterol and zinterol in a rodent LAD-ligation model and found that β2-adrenergic stimulation improved diastolic function. However, we found no evidence of systolic improvements in the clenbu-terol-treated isolated rat hearts.
Mounting evidence strongly implicates that increased myocardial oxidative stress and altered Ca2+ homeostasis play important roles in the myocardial cell injury induced by I/R[3,4,26-28]. Oxidative stress leads to the accumulation of lipid peroxidation product MDA in the heart, which results in cell membrane breakdown and subsequent cell swelling. SOD, as endogenous free radical scavenging enzyme, protects cells against the toxic effects of superoxide radicals[29]. SOD can catalyze the conversion of superoxide radicals to H2O2, which is decomposed into water and oxygen by catalase. It has been demonstrated that reactive oxygen species (ROS) decrease the activity of SR Ca2+-ATPase, a membrane calcium pump that plays an important role in cardiac calcium handling and myocardial relaxation function by coupling cleavage of ATP to transport 2 Ca2+ into the SR lumen. A dysfunction of SR Ca2+-ATPase has been proposed as a contributing factor to MI/R injury[30-33].
In the present study, we first found that clenbuterol increased SOD and decreased MDA levels. In contrast, ICI118551 group had no significant difference from the I/R group. These findings suggest that clenbuterol can attenuate MI/R oxidative stress injury. Our results further demonstrate that clenbuterol significantly increases SR Ca2+-ATPase activity, which might contribute to the improvement of intracellular Ca2+ transients and subsequent relaxation in I/R cardiomyocytes.
It is important to note that apoptosis has been recently proven to represent a dominant form of cardiomyocyte death in I/R[2,27,34]. The inhibition of apoptotic pathways has also been recently shown to improve MI/R injury. There are many ways of detecting apoptotic cell death in situ and in vitro. The TUNEL staining method is now the most widely-used marker for apoptosis in cardiomyocytes. However, it has become clear that TUNEL assay is prone to false positive and negative results[35]. Therefore, the detection of active components of apoptotic pathways, such as caspases and the Bcl family can help to confirm findings[36]. Caspase-3 is considered the central participant of the apoptotic process, and inhibition of its activity retards or even prevents apoptosis[37]. Moreover, the Bcl-2 family plays a key role in the regulation of apoptosis. It is known that Bcl-2 can protect against apoptosis induced by ROS-producing agents, and Bax actively promotes apoptosis[38]. For this reason, we used TUNEL techniques, caspase-3 protein, and Bax/Bcl-2 mRNA expressions to detect apoptosis.
Our results demonstrated that clenbuterol effectively decreased apoptosis via TUNEL staining[39] on I/R injury in isolated rat hearts. Clenbuterol reduced the caspase-3 protein and Bax/Bcl-2 mRNA expressions to inhibit cardio-myocyte apoptosis. Those findings support those of Zhu et al[9] who found that clenbuterol upregulated Bcl-2 and downregulated Bax expressions in the ischemic hippocampus and striatum. These observations strongly suggest that clenbuterol could exert its anti-apoptotic effect on MI/R injury.
A previous study also showed that oxidative stress often caused cell apoptosis. Therefore, we observed the effect of clenbuterol on cultured cardiomyocyte injury and apoptosis induced by H2O2 in vitro. H2O2, a major kind of ROS, can lead to the formation of hydroxyl radicals mediated by intracellular heavy metal ions through the Fenton reaction. Both H2O2 and hydroxyl radicals induce severe intracellular oxidative stress, which eventually lead to apoptosis and necrosis of cardiomyocytes[39-40]. In our study, when cardiomyocytes were exposed to exogenous H2O2, cell viability decreased and apoptosis increased significantly. Pretreatment with 1 µmol/L clenbuterol greatly increased cell viability and decreased apoptosis induced by H2O2. These results suggest that clenbuterol has protective effects in H2O2-induced apoptosis.
In conclusion, the present results provide evidence that β2-adrenergic agonist clenbuterol significantly improves diastolic function (LVEDP and CF), increases SOD and Ca2+-ATPase activity, and decreases the MDA level and LDH release. Our data also show that clenbuterol could inhibit MI/R-induced apoptosis in isolated rat hearts. Moreover, clenbuterol protects cultured cardiomyocytes from H2O2-induced injury and decreases apoptosis in vitro. These findings provide a novel insight into the role of clenbuterol in improving ventricular diastolic function and decreasing oxidative stress and apoptosis on I/R injury in isolated rat hearts. Clenbuterol also decreases apoptosis induced by H2O2.
However, further studies will investigate whether these effects of clenbuterol treatment on isolated rat hearts have time-dependence and dose-dependence. In addition, the mechanisms by which clenbuterol attenuates MI/R injury were not elucidated by this study. Improvements in ventricular diastolic function, oxidative stress, and apoptosis are only implicated as possible mechanisms. Future studies will better characterize the effects and pathways of β2-adrenergic stimulation in the I/R heart.
Author contribution
Qing FU, Ping LIU designed research; Ping LIU, Lei ZHAO, Ben-rong HU, Lei YANG performed research; Qin FU, Ji-zhou XIANG contributed new reagents and analytical tools; Ping LIU analyzed the data; Qing FU, Ji-zhou XIANG, Ping LIU wrote the paper.
References
- Gill S, Haastrup B, Haghfelt T, Dellborg M, Clemmensen P. Continuous vectorcardiography is superior to standard electrocardiography in the prediction of long-term outcome after thrombolysis in patients with acute myocardial infarction. Coron Artery Dis 2002;13:169-75.
- Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischemia and infarction. J Clin Pathol 2002;55:801-11.
- Kumer D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med 2003;142:288-97.
- Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 2001;89:279-86.
- Salorinne Y, Stenius B, Tukiainen P, Poppius H. Double-blind cross-over comparison of clenbuterol and salbutamol tablets in asthmatic out-patients. Eur J Clin Pharmacol 1975;8:189-95.
- Choo JJ, Horan MA, Little RA, Rothwell NJ. Anabolic effects of clenbuterol on skeletal muscle are mediated by beta 2-adrenoceptor activation. Am J Physiol 1992;263:50-6.
- Maltin CA, Delday MI, Watson JS, Heys SD, Nevison IM, Ritchie IK, et al. Clenbuterol, a beta-adrenoceptor agonist, increases relative muscle strength in orthopaedic patients. Clin Sci (Lond) 1993;84:651-4.
- Xydas S, Kherani AR, Chang JS, Klotz S, Hay I, Mutrie CJ, et al. beta(2)-Adrenergic stimulation attenuates left ventricular remodeling, decreases apoptosis, and improves calcium homeostasis in a rodent model of ischemic cardiomyopathy. J Pharmacol Exp Ther 2006;317:553-61.
- Zhu Y, Prehn JH, Culmsee C, Krieglstein J. The beta2-adrenoceptor agonist clenbuterol modulates Bcl-2, Bcl-xl and Bax protein expression following transient forebrain ischemia. Neuroscience 1999;90:1255-63.
- Zhu Y, Culmsee C, Roth-Eichhorn S, Krieglstein J. Beta(2)-adrenoceptor stimulation enhances latent transforming growth factor-beta-binding protein-1 and transforming growth factor-beta 1 expression in rat hippocampus after transient forebrain ischemia. Neuroscience 2001;107:593-602.
- Culmsee C, Semkova I, Krieglstein J. NGF mediates the neuroprotective effect of the beta2-adrenoceptor agonist clenbuterol in vitro and in vivo: evidence from an NGF-antisense study. Neurochem Int 1999;35:47-57.
- Culmsee C, Stumm RK, Schafer MK, Weihe E, Krieglstein J. Clenbuterol induces growth factor mRNA, activates astrocytes and protects rat brain tissue against ischemic damage. Eur J Pharmacol 1999;379:33-45.
- Ponicke K, Groner F, Heinroth-Hoffmann I, Brodde OE. Agonist-specific activation of the beta2-adrenoceptor/Gs-protein and beta2-adrenoceptor/Gi-protein pathway in adult rat ventricular cardiomyocytes. Br J Pharmacol 2006;147:714-9.
- Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, et al. The beta (2) adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3’-kinase. Circ Res 2000;87:1172-9.
- Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, Raynolds MV, Sastravaha A, et al. Myocardial-directed overexpression of the human beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol 2000;32:817-30.
- Steinberg SF. The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res 1999;85:1101-11.
- Clark LA, Taylor PM, Brand N. Stimulation of growth and collagen secretion in cardiac fibroblasts by clenbuterol. J Mol Cell Cardiol 2006;40:947-54.
- George I, Xydas S, Mancini DM, Lamanca J, DiTullio M, Marboe CC, et al. Effect of clenbuterol on cardiac and skeletal muscle function during left ventricular assist device support. J Heart Lung Transplant 2006;25:1084-90.
- Martinek RG. A rapid ultraviolet spectrophotometric lactic dehydrogenase assay. Clin Chim Acta 1972;40:91-9.
- Xu D, Li Y, Wang J, Davey AK, Zhang S, Evans AM. The cardioprotective effect of isosteviol on rats with heart ischemia–reperfusion injury. Life Sci 2007;80:269-74.
- Jones LR. Mg2+ and ATP effects on K+ activation of the Ca2+-transport ATPase of cardiac sarcoplasmic reticulum. Biochim Biophys Acta 1979;557:230-42.
- Kodavanti PR, Cameron JA, Yallapragada PR, Desaiah D. Effect of chlordecone (kepone) on calcium transport mechanisms in rat heart sarcoplasmic reticulum. Pharmacol Toxicol 1990;67:227-34.
- Pande M, Cameron JA, Vig PJ, Desaiah D. Phencyclidine block of Ca2+-ATPase in rat heart sarcoplasmic reticulum. Toxicology 1998;129:95-102.
- Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. Beta-adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem 1983;258:464-71.
- Ahmet I, Krawczyk M, Heller P, Moon C, Lakatta EG, Talan MI. Beneficial effects of chronic pharmacological manipulation of beta-adrenoreceptor subtype signaling in rodent dilated ischemic cardiomyopathy. Circulation 2004;110:1083-90.
- Kalsia KK, Soppaa GKR, Terraccianoa CMN, Smolenskia RT, Yacouba MH. Oxidative capacity recovers in the unloaded heart with clenbuterol. J Mol Cell Cardiol 2006;40:933-40.
- Das Dk Maulik N. Antioxidant effectiveness in ischemia-reperfusion tissue injury. Methods Enzymol 1994;233:601-10.
- Ferdinandy P, Schulz R. Nitric oxide, superoxide and peroxynitrite in myocardial ischemia-reperfusion injury and preconditioning. Br J Pharmacol 2003;138:532-43.
- Cordis GA, Maulik N, Bagchi D, Engelman RM, Das DK. Estimation of the extent of lipid peroxidation in the ischemic and reperfused heart by monitoring lipid metabolic produces with the aid of high-performance liquid chromatography. J Chromatogr 1993;632:97-103.
- Kaneko M, Beamish RE, Dhalla NS. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am J Physiol 1989;256:368-74.
- Ikemoto N. Structure and function of the calcium pump protein of sarcoplasmic reticulum. Annu Rev Physiol 1982;44:297-317.
- Inesi G. Mechanism of calcium transport. Annu Rev Physiol 1985;47:573-601.
- Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 1997;49:1-51.
- Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res 1996;79:949-56.
- Aigner T. Apoptosis, necrosis, or whatever: how to find out what really happens? J Pathol 2002;198:1-4.
- Kanoh M, Takemura G, Misao J, Hayakawa Y, Aoyama T, Nishigaki K, et al. Significance of myocytes with positive DNA in situ nick end-labeling (TUNEL) in hearts with dilated cardiomyopathy: not apoptosis but DNA repair. Circulation 1999;99:2757-64.
- Sulpizi M, Rothlisberger U, Carloni P. Molecular dynamics studies of caspase-3. Biophys J 2003;84:2207-15.
- Delchev SD, Georgieva KN, Koeva YA, Atanassova PK. Bcl-2/Bax ratio, mitochondrial membranes and aerobic enzyme activity in cardiomyocytes of rats after submaximal training. Folia Med (Plovdiv) 2006;48:50-6.
- Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G, et al. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 1999;99:3071-8.
- von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 1999;99:2934-41.