
Identification of antiviral mimetic peptides with interferon α-2b-like activity from a random peptide library using a novel functional biopanning method1
Introduction
Phage display has been proven as a highly effective approach for designing cytokine variants and developing small molecular drugs[1]. As evidenced by numerous studies within the past decade, the extracellular domain of cytokine receptors can be mapped by screening peptide libraries, and small mimetic peptides corresponding to the active binding area of their cognate cytokines can be consequently identified and developed as macromolecular drug substitutes[2]. Wrighton et al isolated a 14 amino acid peptide targeted to the erythropoietin (EPO) cytokine receptor. This small peptide specifically bound the EPO receptor and activated the appropriate signaling pathways, while also retaining biological activity[3]. Likewise, Cwirla et al identified a 14 amino acid peptide from recombinant peptide libraries. This peptide displayed high affinity to the human thrombopoietin (TPO) receptor and stimulated the proliferation of a TPO-responsive Ba/F3 cell line[4]. Another group used human CD81 (hCD81)-expressing murine NIH/3T3 cells to select a hCD81-binding peptide from a phage-display nonapeptide library. The motif-containing phages were able to competitively inhibit the binding of hepatitis C virus enveloping glycoprotein 2 to native hCD81-expressing MOLT-4 cells[5]. In the same manner, many mimetic peptides of various cytokines have been harvested, including melanocytes-stimulating hormone (MC)[6], estrogen[7], interleukins (IL), and TNF[8,9].
Interferon (IFN) is currently the human protein most widely used as a therapeutic agent, having been approved to treat various types of viral diseases. However, its efficient treatment is often hampered by various side-effects, such as neutralizing antibodies and cell resistance[10,11]. In overcoming these difficulties, the mimetic or synthetic peptide approach is very attractive. For instance, Sato et al described the successful mimicry of IFN-β by a peptide isolated from phage-display screening using a neutralizing anti-IFN-β monoclonal antibody. Despite the lack of similarity to the primary sequence of IFN-β, this 15-mer peptide retained the ability to bind the type I IFN receptor, as well as the antiviral activity of IFN-β[12]. Based on a similar strategy, Hu et al selected 2 peptides using a neutralizing antibody from a phage-display hexapeptide library; one of these peptides (WLDPRH) exhibited structural homology with loop AB (29–35), the binding domain of the type I IFN receptor. Although it exhibited no activity alone, this peptide was evidently able to cooperatively improve the antiviral activity of IFN-α[13]. In yet another example, a synthetic peptide corresponding to residues 95–133 of murine IFN-γ was shown to possess antiviral activity[14].
In fact, it should be a straightforward strategy to obtain agonist peptides that bind to and stimulate the IFN receptor (IFNAR) by screening a phage-display library with the IFN receptor. However, there are some technical difficulties in the purification and maintenance of the native structure when working with membrane receptors. Previous studies have shown that screening with a soluble form of the type I IFN receptor failed to identify any peptide ligands[12]. Therefore, it appears better to use whole native or transfected cells to select ligands[15,16]. Whole cells usually produce receptors that maintain the native conformation with normal post-translational modification, so ligands can be selected even without detailed information on the nature of the receptor[5]. However, there have been few reports of IFN mimetic peptides possessing antiviral activity being discovered by direct cell-based selection. In a previous study[17], we performed 4 rounds of biopanning against intact native WISH cells expressing IFNα-2b receptors, followed by 3 rounds of selection with polyclonal anti-IFNα-2b antibodies. This approach resulted in the enrichment of phage clones binding both to IFNα-2b receptors and anti-IFNα-2b antibodies. Unfortunately, almost all of the phage clones randomly picked out from the last round of selection were antagonist peptides of IFNα-2b. In the current study, we developed a functional selection strategy to obtain antiviral mimetic peptides of IFNα-2b in the hopes of designing IFN mimetic peptide agonists that bind with higher affinity to the receptor.
Materials and methods
Reagents IFNα-2b standard (1.0×1011 IU/g IFN protein) was kindly provided by Tianjin Hualida Bioengineering (Tianjin, China). Cell culture media and reagents were obtained from Gibco BRL Life Technologies (Gaithersburg, MD, USA). The phage-display heptapeptide (PhD-7) library kit (1.5×1016 plaque-forming units [pfu]/L) was purchased from New England Biolabs (Beverly, MA, USA). Wild-type M13 phage and the horseradish peroxidase (HRP)-conjugated anti-M13 phage antibody were purchased from Pharmacia Biotech (Uppsala, Sweden). The fusion protein GFP/IFNα-2b was prepared as outlined in our previous studies[17]. Other chemicals used in this study were of analytical grade and were commercially available.
Cell culture WISH cells and vesicular stomatitis virus (VSV) were kindly provided by Hualida Bioengineering. The WISH cells, which endogenously express type I IFN receptors on the cell surface, were cultured at 37 °C with 5% CO2 in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 1×105 U/L penicillin, and 0.1 g/L streptomycin.
Functional selection The primary PhD-7 library, which had been previously subjected to 4 rounds of biopanning against WISH cells as described[18], was used for the functional selection. The selection procedure was based on combining the PhD-7 library kit standard procedure with some modifications for functional screening for the protection of human amnion WISH cells against VSV-induced cytopathic effects[18]. WISH cells (2.0×104) were seeded into 96-well culture plates and incubated at 37 °C for 3 h. Approximately 1.0×1011 pfu phage were transferred to the 96-well culture plates and incubated at 37 °C for 24 h. The cells were then challenged with VSV and incubated at 37 °C for an additional 24 h. After washing 10 times with RPMI-1640 medium and 5 times with phosphate-buffered saline (PBS; 0.02 mol/L sodium phosphate buffer, pH 7.4, and 0.15 mol/L NaCl); the remaining phages that had bound to surviving cells were eluted for 30 min with 0.01 g/L IFNα-2b. The eluted phages were replicated by infecting Escherichia coli ER2738 cells. The amplified phage particles were purified using polyethylene glycol and then used for subsequent rounds of selection.
Phage ELISA Phage ELISA was performed according to our previous report[17]. Briefly, approximately 1.0×1010 pfu-amplified phages (or mixed with 0.01 g/L IFNα-2b) were incubated with WISH cells (2.0×105 cells/well) fixed onto 96-well culture plates with glutaraldehyde. After extensive washing with PBST (PBS containing 0.1% Tween 20), the bound phages were detected with the HRP-conjugated anti-M13 phage antibody. Wild-type M13 phage was used as a negative control.
Peptide sequencing and synthesis The single-stranded DNA (ssDNA) was prepared from identified phage clones as described in the PhD-7 library kit guidelines and sequenced by the Shanghai Sangon Company (Shanghai, China). Corresponding amino acid sequences were deduced from DNA sequences; 2 peptides corresponding to positive clones T3 and T9 and their corresponding IFN sequences were synthesized chemically by GL Biochem (Shanghai, China) and designated as IR-7 (clone T3, IRPDTPR), VR-7 (IFN143-149, VRAEIMR), KP-7 (clone T9, KNVHPPP) and KG-7 (IFN31-37, KDRHDFG). Two antagonist peptides obtained from previous studies[17], SP-7(SLSPGLP) and FY-7(FSAPVRY), were also used as the controls. At the same time, the 4 mutant peptides of T3 and T9 were synthesized and used in antiviral activity assay, and designated as IR-7A (IGPDTPR), IR-7B (IRPDTPG), KP-7A (GNVHPPP) and KP-7A (KNVGPPP). The key residues, Arg144 or Arg149 of IR-7 and Lys31 or His34 of IR-7, were replaced with Gly, respectively.
Synthetic peptide competition assay Exponentially-growing WISH cells were fixed on culture plates using glutaraldehyde (2.0×105 cells/well). After blocking with PBS (containing 1% bovine serum albumin and 0.1% NaN3) for 1 h at room temperature, 1 µg GFP/IFNα-2b was added to each well and incubated at 37 °C for 2 h. After washing 5 times with PBST, different amounts of synthetic peptides were added and incubated at 37 °C for 1 h with gentle shaking. The wells were then washed with PBST again and observed with an inverted fluorescence microscope. The intensity of fluorescence was analyzed with Image Pro Plus 5.1 and the IC50 values were calculated with GraphPad Prism 4 software[19].
Antiviral activity assay The antiviral activity of IFN and synthetic peptides was determined in vitro by the protection of human amnion WISH cells against VSV-induced cytopathic effects, as described for the traditional method[18]. In brief, 2.0×104 WISH cells were seeded into each well of 96-well plates and incubated with samples for 24 h at 37 °C. After incubation, the cells were challenged with VSV and the plates were incubated at 37 °C for 24 h. Virus-induced cytopathic effects were assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method[20].
Results
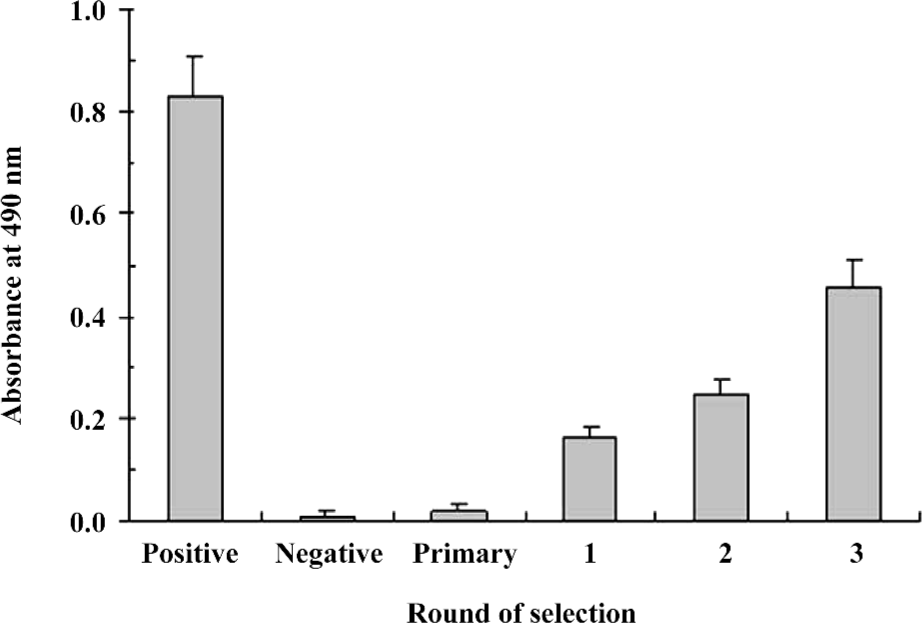
Specific enrichment of positive phages with antiviral activity In order to efficiently enrich IFNAR-binding phages with functional antiviral activity, we chose a phage-display library featuring specific binding to IFNAR as the primary library for further functional selection. The functional enrichment was determined by the antiviral activity after each round of selection. In 3 rounds of functional selection, the antiviral activity increased markedly after each round compared to the primary library (Figure 1), indicating enrichment for positive phages with antiviral activity.
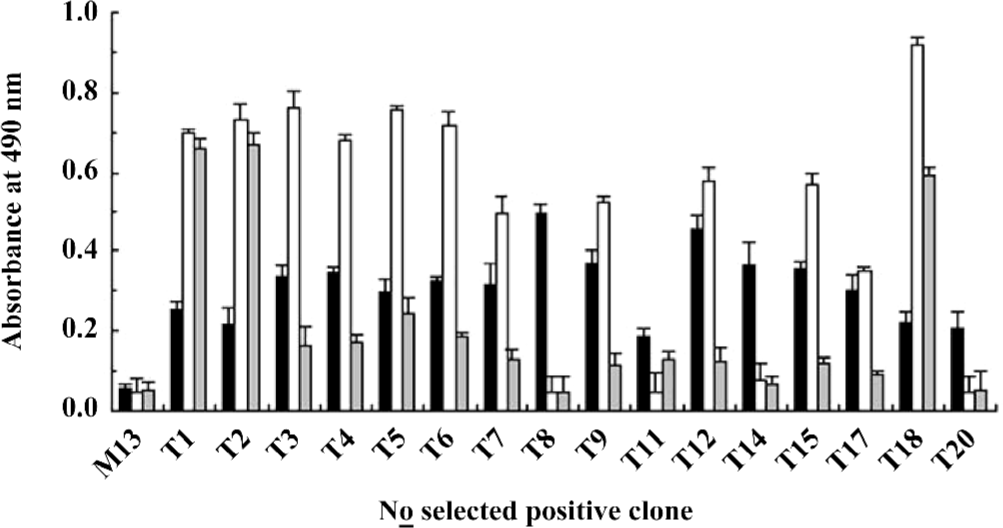
Identification of the positive phages Clones (1012) were picked out from the sample after the third round of functional selection and the antiviral activity was examined. Only 16 clones (approximately 1.6%) showed antiviral activity (Figure 2). Further phage ELISA testing demonstrated that 12 out of the 16 positive clones (T1–9, 11, 12, 14, 15, 17, 18, and 20) could bind to WISH cells, and 10 out of these 12 binding clones (T3–7, 9, 12, 15, 17, and 18) could be specifically competed off by 1 µg IFNα-2b (Figure 2). These results confirmed the success of the functional selection scheme in enriching IFNAR-binding phages with antiviral activity.
Analyses of exogenous sequences of positive-phage clones The ssDNA prepared from the 10 positive clones were sequenced, and the amino acid sequences of the mimetic peptides were then deduced from the DNA sequences. A homologous analysis of amino acid sequences revealed that the selected clones were corresponding to 7 domains defined by residues of IFNα-2b (T9: AB loop 31–37; T18: BC loop 68–74; T4: C helix 93–99; T6, T17: CD loop 105–112; T12: D helix 115–121; T5: DE loop 132–138 and T3, T7, T15: E helix 143–161; Table 1). It has been proposed that the AB loop (residues 26–35) and E helix (residues 144–153) mediate the interaction with receptors[13]. By comparing the homology with the amino acid sequence of the known functional domain in IFNα-2b, 2 peptides represented by the positive clones T3 and T9 and aligned with the IFNAR2-binding domains (AB loop and E helix), their mutant peptides, and their corresponding native IFN sequences were synthesized and designated IR-7, KP-7, IR-7A, IR-7B, KP-7A, KP-7B, VR-7, and KG-7, respectively.

Full table
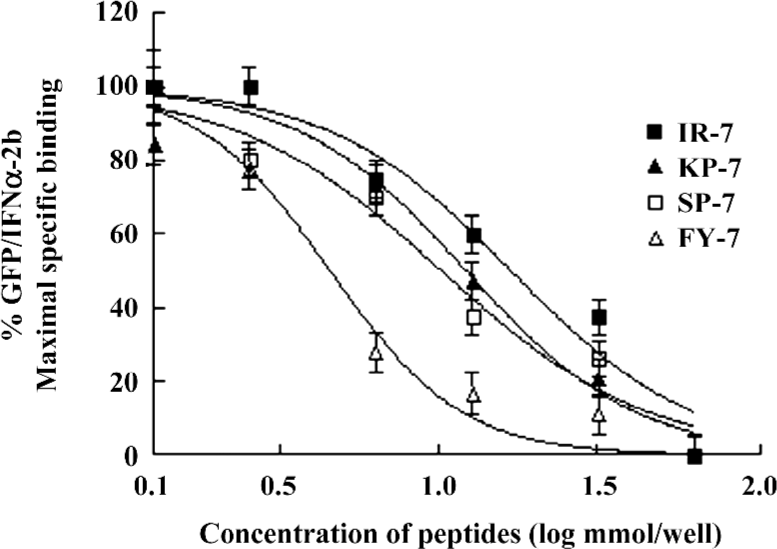
Competition reactivity of mimetic peptides to IFNAR The abilities of 2 mimetic peptides, IR-7 and KP-7, to inhibit the binding of GFP/IFNα-2b to IFNAR were determined by a competitive assay. As shown in Figure 3, IR-7 and KP-7 reduced fluorescence-bound GFP/IFNα-2b in a dose-dependent manner as well as 2 antagonist peptides, SP-7 and FY-7, reported previously[17]. The IC50 values of IR-7 and KP-7 were 16.8 and 11.9 nmol, respectively, slightly higher than SP-7 (11.2 nmol) and FY-7 (4.03 nmol). These results confirmed that 2 mimetic peptides did effectively mimic IFNα-2b and inhibited the binding of GFP/IFNα-2b to IFNAR.
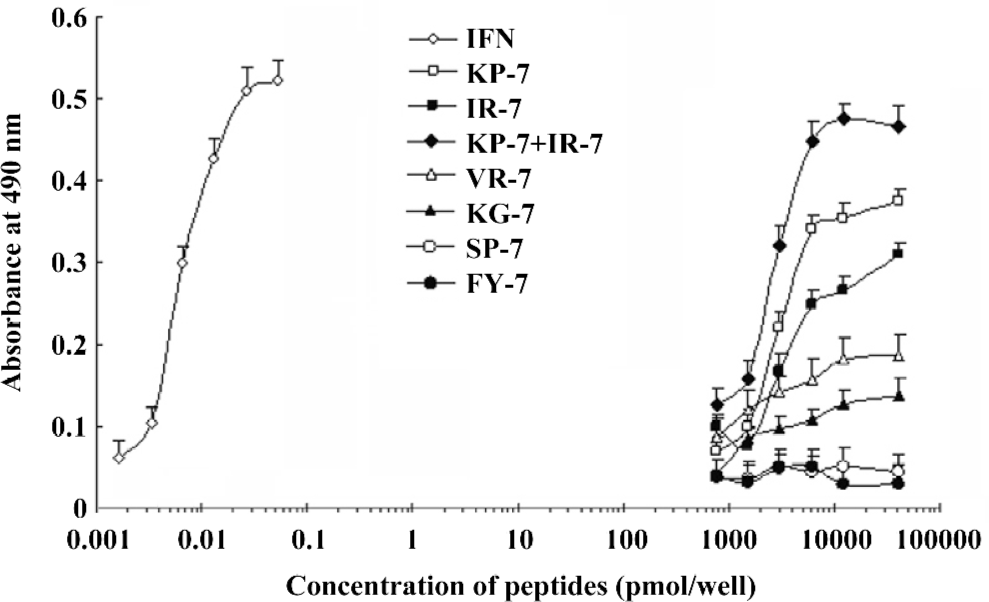
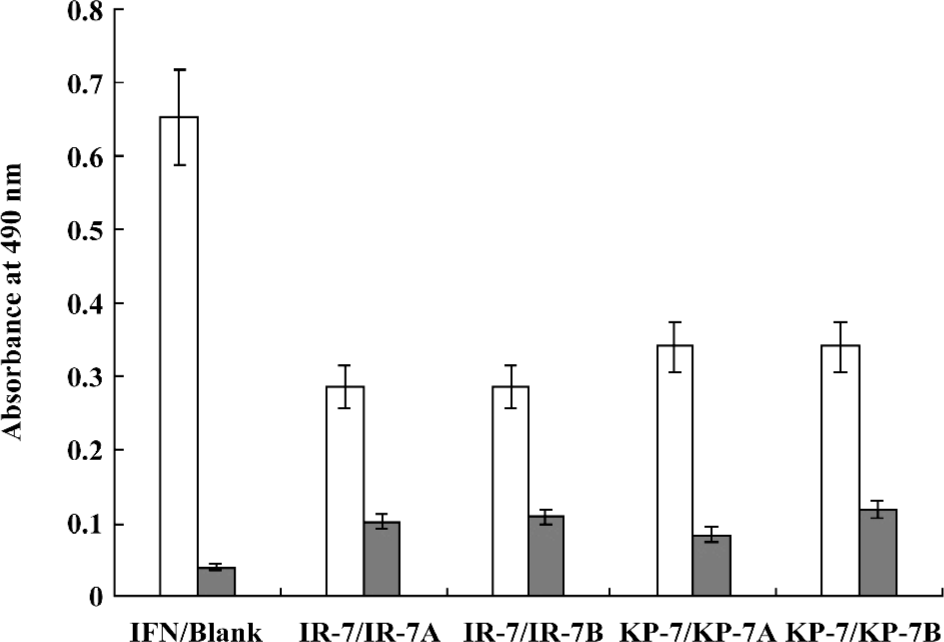
Antiviral activity of mimetic peptides The antiviral activity of the mimetic peptides was determined by the MTT method. When the peptide was added with an amount between 1.5 and 6.0 nmol, both mimetic peptides (IR-7 and KP-7) showed significant antiviral activity in a dose-dependent manner compared to their corresponding IFN sequences peptides (VR-7 and KG-7) and the antagonist control peptides (SP-7 and FY-7; Figure 4), which suggested that the secondary structure of peptides was very important in antiviral activity. At the same time, these experiments indicated that the antiviral activity of KP-7 was slightly stronger than that of IR-7. Furthermore, the antiviral activity of a KP-7 and IR-7 mixture was even stronger still than that of KP-7 or IR-7 alone at the same dosage, indicating that the 2 domains, the AB loop (residues 31–37) and E helix (residues 143–149), in IFNα-2b can inhibit viral infection cooperatively. Comparing IR-7 or KP-7 with its mutant peptides, the antiviral activity of original mimetic peptides was obviously stronger than its mutant peptides (Figure 5), and this result suggested that the key residue was indispensable.
Discussion
Type I IFN is a family of homologous cytokines that potently elicits an antiviral and antiproliferative state in cells. All human type I IFN (IFN-α, -β, -τ, and -ω) bind to a cell surface receptor consisting of 2 transmembrane subunits, IFNAR1 and IFNAR2, that associate on binding. The binding of IFN to its receptor triggers a cascade of events, activating a number of proteins that inhibit viral replication and cell growth[21].
In previous attempts to develop antagonist peptides to IFNα-2b, we performed biopanning against WISH cells and polyclonal anti-IFN antibodies. The method provided a final-phage display library in which the phage clones could bind both to IFN receptors and anti-IFN antibodies[17]. However, many of these clones might not activate the receptors to stimulate the IFN signaling pathway. Building on these tactics, we developed a third strategy called “functional biopanning selection” in order to obtain mimetic peptides of IFNα-2b with antiviral activity. In this functional selection procedure, VSV was used to infect WISH cells that had been treated with the phage-display library. After incubation, most of the infected WISH cells had died and could be washed away together with phage clones without antiviral activity. However, on the surface of the surviving cells, we anticipated that there might be combined phage clones possessing protective and antiviral activity, and that these candidates could be competitively eluted by IFNα-2b. Our results confirmed that positive phages with antiviral activity had indeed been enriched after 3 rounds of functional panning selection (Figure 1).
Although Figure 1 shows that the survival of WISH cells in the third round was approximately 60% compared with the positive control, the positive hit rate was only 1.6%. One explanation for this phenomenon is that there are plenty of IFN receptors on the surface of WISH cells, but the activation of only a few can result in antiviral activities[22]. Therefore, most of phage clones synchronously bound to IFN receptors on surviving WISH cells during the functional selection, but some binding was irrelative with antiviral activity. Among the 12 positive phage clones selected on WISH cells, 10 clones could bind to IFNAR specifically (T3–7, 9, 12, 15, 17, and 18), so the antiviral activities of these clones should be related to the IFN signaling pathway.
It has been recently determined by X-ray crystallography that the IFNα-2b molecule is composed of 5 α-helices (A–E) linked by 1 long connection (AB loop) and 3 short segments (the BC, CD, and DE loops)[23]. In this study, the sequences of 10 positive clones were corresponding to 7 domains defined by residues of IFNα-2b (T9: AB loop 31–37; T18: BC loop 68–74; T4: C helix 93–99; T6, T17: CD loop 105–112; T12: D helix 115–121; T5: DE loop 132–138 and T3, T7, T15: E helix 143–161; Table 1). The A helix (residues 12–15), the AB loop (residues 26–35), and the E helix (residues 144–153) of IFN-α binds IFNAR2, triggering the induction of antiviral activity[13,24]. Therefore, it is possible that the peptides of T9 are structural mimics of conformational epitopes of the IFN AB loop, and those of T3, T7, and T15 might be E helix mimics.
Mitsui et al and Uzé et al’s studies revealed that if the amino acid sites of Leu30, Lys31, Arg33, His34, Phe36, Arg120, Lys121, Tyr122,Gln124, Tyr129, Lys131, and Glu132 on IFNα are mutated, the bioactivity is observably reduced[25,26]. This indicates that these sites are important for IFN-introduced antiviral activity; however, Lys31 and His34 of T9 lie within this range. In Jacob’s research, 4 conservative amino acids, Arg144, Ala145, Met148, and Arg149 in the E helix were identified as “hot amino acids” key points for the interaction between IFN and its receptor[21,27]. Clone T3 includes Arg144 and Arg149. In addition, similarity analysis indicated that Glu146 in IFN and Asp146 of T3 were equivalent, since they are both acidic amino acids. We thus selected clones T9 and T3 for peptide synthesis, designated as KP-7 and IR-7. These 2 peptides were not only capable of specific binding to the IFN receptor (Figure 3), but also provided antiviral activity in a dose-dependent manner (Figure 4). Furthermore, we observed a cooperative effect on antiviral activity when both peptides were added simultaneously. To confirm the function of key residues from mimetic peptides, we synthesized 4 mutant peptides at key residue (Lys31 and His34 of T9; Arg144 and Arg149 of T3) and performed an antiviral activity assay. The results indicated again that the key points were indispensable in the IFN antiviral process.
In this study, we completed only preliminary studies for T3 and T9, although other positive clones also had some antiviral activity. For example, T4 (93–99) partially overlaps with the C helix, which is involved in the binding of IFN to IFNAR1[28]. In addition, Ser68 of T18, Arg120 of T12, and Lys133 of T5 are known to be important amino acids for IFN bioactivity. Therefore, these clones also hold interest for further studies.
Using cell selection and functional biopanning, 2 IFNα-2b agonist peptides with antiviral activity, KP-7 and IR-7, were successfully selected from a random phage-display library. The results of this novel functional biopanning technique suggest that IFNAR-induced antiviral activity may require multiple binding domains, an idea that may prove helpful in further elucidating the molecular mechanism of IFN receptor binding. Moreover, this functional biopanning technique should be useful in the development of mimetic peptide drugs of human IFN and other cytokines.
References
- Schooltink H, Rose-John S. Designing cytokine variants by phage-display. Comb Chem High Throughput Screen 2005;8:173-9.
- Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W. Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev 2006;58:1622-54.
- Wrighton NC, Farrell FX, Chang R, Kashyap AK, Barbone FP, Mulcahy LS, . Small peptides as potent mimetics of the protein hormone erythropoietin. Science 1996; 273: 46 458–87.
- Cwirla SE, Balasubramanian P, Duffin DJ., Wagstrom CR, Gates CM, Singer SC, et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science 1997;276:1696-9.
- Cao J, Zhao P, Miao XH, Zhao LJ, Xue LJ, Qi ZT. Phage display selection on whole cells yields a small peptide specific for HCV receptor human CD81. Cell Res 2003;13:473-9.
- Szardenings M, Muceniece R, Mutule I, Mutulis F, Wikberg JE. New highly specific agonistic peptides for human melanocortin MC(1) receptor. Peptides 2000;21:239-43.
- Venkatesh N, Zaltsman Y, Somjen D, Gayer B, Boopathi E, Kasher R, et al. A synthetic peptide with estrogen-like activity derived from a phage-display peptide library. Peptides 2002;23:573-80.
- Doorbar J, Winter G. Isolation of a peptide antagonist to the thrombin receptor using phage display. J Mol Biol 1994;244:361-9.
- Kimura T, Kaburaki H, Miyamoto S, Katayama J., Watanabe Y. Discovery of a novel thrombopoietin mimic agonist peptide. J Biochem (Tokyo) 1997;122:1046-51.
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001;14:778-809.
- Schellekens H, Casadevall N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol 2004;251:114-9.
- Sato A, Sone S. A peptide mimetic of human interferon (IFN)-β Biochem J 2003;371:603-8.
- Hu R, Ma XJ, Wei KK, Wang H, Qian ZC, Hou YD. Influence of the synthetic peptide recognized by neutralizing antibody on IFN-induced biological responses Chinese J Exp Clin Virol 1999;13:29-32.
- Ahmed CM, Burkhart MA, Subramaniam PS, Mujtaba MG, Johnson HM. Peptide mimetics of γ-interferon possess antiviral properties against vaccinia virus and other viruses in the presence of poxvirus B8R protein. J Virol 2005;79:5632-9.
- Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, et al. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J 2000;19:1525-33.
- Szardenings M, Tornroth S, Mutulis F, Ruta M, Kari K, Arja K, . Phage display selection on whole cells yields a peptide specific for melanocortin receptor 1. J Biol Chem 1997; 272: 27 943–8.
- Tian W, Bai G, Li ZH, Yang WB. Antagonist peptides of human interferon-α2b isolated from phage display library inhibit interferon induced antiviral activity. Acta Pharmacol Sin 2006;37:1044-50.
- Jacob P, Gideon S. Biophysical analysis of the interaction of human ifnar2 expressed in E coli with IFNα2. J Mol Biol 1999;289:57-67.
- Chen JQ, Bai G, Yang Y, Geng P, Cao Y, Zhu YY. Identifying glucagon-like peptide-1 mimetics using a novel functional reporter gene high-throughput screening assay. Peptides 2007;28:928-34.
- Abe K, Matsuki N. Measurement of cellular 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reduction activity and lactate dehydrogenase release using MTT. Neurosci Res 2000;38:325-9.
- Chill JH, Quadt S R, Levy R. The human type I interferon receptor: NMR structure reveals the molecular basis of ligand binding. Structure (Camb) 2003;11:791-802.
- Ma XJ, Hu R, Lu H, Wei KK, Zhang LL, Xue SX, et al. Engineering human interferon α1c/86D with phage display technology. Science in China Ser. C 1999;29:209-16.
- Ramaswamy R, Leigh JW, Alan H, Paul R, Paul PT, Tattanahalli LN, et al. Zinc mediated dimmer of human interferon-α2b revealed by X-ray crystallography. Structure 1996;4:1453-63.
- Roisman LC, Piehler J, Trosset JY, Scheraga HA, Schreiber G. Structure of the interferon-receptor complex determined by distance constraints from double-mutant cycles and flexible docking. Proc Natl Acad Sci USA 2001;98:13231-6.
- Mitsui Y, Senda T, Shimazu T. Structural, functional and evolutionary implications of the three-dimensional crystal structure of murine interferon-β. Pharmacol Ther 1993;58:93-132.
- Uzé G, Lutfalla G, Mogensen KE. Alpha and beta interferons and their receptor and their friends and relations. J Interferon Cytokine Res 1995;15:3-26.
- Piehler J, Roisman LC, Schreiber G. New structure and functional aspects of the type I interferon-receptor interaction revealed by comprehensive mutational analysis of the binding interface. J Biol Chem 2000;275:40425-33.
- Seto MH, Harkins RN, Adler M. Homology model of human interferon-α8 and its receptor complex. Protein Sci 1995;4:655-70.